Болезнь Помпе (ген GAA) (Pompe disease (GAA gene))
Метод определения Секвенирование по Сэнгеру.
Исследуемый материал Кровь (ЭДТА)
Синонимы: Гликогеноз II типа, ген GAA.
Pompe disease, mutations in GAA gene.
Краткое описание исследования «Болезнь Помпе (ген GAA)»
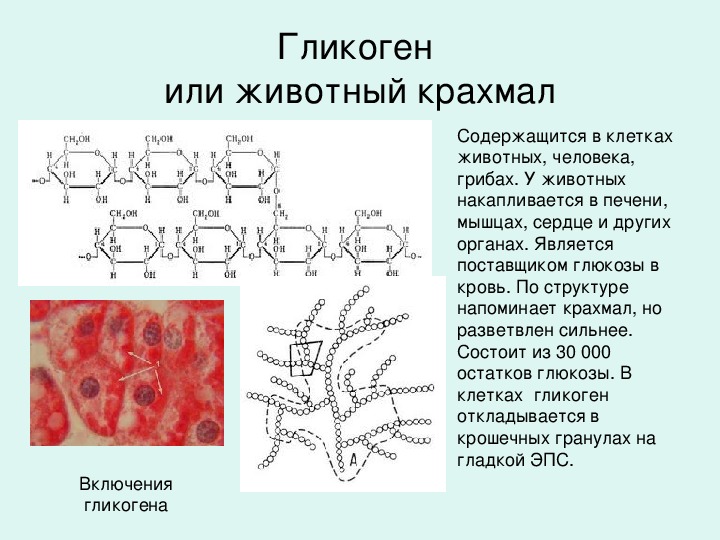
Болезнь Помпе (гликогеноз II типа) — прогрессирующее аутосомно-рецессивное заболевание, обусловленное аберрантным накоплением гликогена в органах и тканях.
Ген GAA кодирует фермент кислую альфа-глюкозидазу, участвующую в расщеплении гликогена лизосомами. Гликоген является универсальным способом накопления и хранения углеводов в организме человека. В норме излишки углеводов, поступающих с пищей, запасаются в различных органах и тканях (преимущественно в мышцах и печени) в виде гликогена и по мере необходимости высвобождаются из депо. При болезни Помпе процесс высвобождения энергии из гликогена останавливается, что обуславливает с одной стороны дефицит питательных веществ в организме, а с другой стороны — патологическое увеличение и нарушение функции органов, в которых в норме накапливается гликоген.
Выделяют младенческую форму заболевания и болезнь Помпе с поздним началом. Возраст дебюта, скорость прогрессии и тяжесть проявлений обусловлены видом аберраций, обнаруженных в гене GAA.
Младенческая форм болезни Помпе дебютирует в первые месяцы жизни. Обращает на себя внимание выраженная гипотония мышц с прогрессирующей мышечной слабостью. Такие дети быстро устают при грудном кормлении, малоподвижны, отстают в росте и психомоторном развитии. Нередко отмечается поражение сердечной мышцы и ослабление дыхания, способные представить значительную угрозу жизни младенца.
Более мягкие формы могут провялятся в более позднем детском возрасте. В этом случае на себя внимание обращают сложности в различных видах двигательной активности, занятиях спортом, быстрая утомляемость, отставание в росте и развитии, увеличение внутренних органов. По данным лабораторных показателей можно обнаружить повышение сывороточной креатинфосфокиназы.
Болезнь Помпе с поздним дебютом характеризуется наследованием «мягких» патогенных вариантов в гене GAA и высокой гетерогенностью клинических проявления, составляющих существенную сложность для дифференциальной диагностики.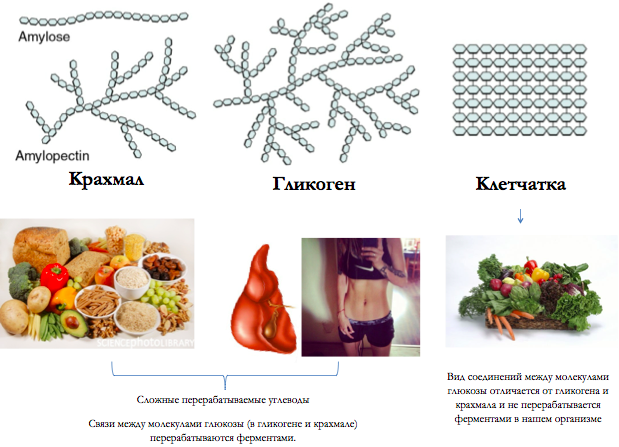 Основной жалобой таких пациентов является слабость мышц различной выраженности при занятиях спортом и интенсивной физической активности. Нередко обследуемые жалуются на сложности при вставании с постели, поднятия по лестнице. Со временем состояние прогрессирует, и к нему присоединяется дыхательная недостаточность, кардиомегалия, выраженная слабость. Для замедления прогрессирования заболевания применяется ферментная заместительная терапия при верификации диагноза молекулярно-генетическими метолами.
Основной жалобой таких пациентов является слабость мышц различной выраженности при занятиях спортом и интенсивной физической активности. Нередко обследуемые жалуются на сложности при вставании с постели, поднятия по лестнице. Со временем состояние прогрессирует, и к нему присоединяется дыхательная недостаточность, кардиомегалия, выраженная слабость. Для замедления прогрессирования заболевания применяется ферментная заместительная терапия при верификации диагноза молекулярно-генетическими метолами.
С какой целью выполняют исследование «Болезнь Помпе (ген GAA)»
Тест предназначен для диагностики болезни Помпе.
как начинается редкая болезнь Помпе и как ее лечат
Изображение: Freepik
Редкая патология встречается у одного человека из 14 000-300 000.
15 апреля во всем мире отмечается день осведомленности о редком нервно-мышечном заболевании — болезни Помпе, которую также называют болезнью накопления гликогена II типа. Она развивается как у детей, так и у взрослых, вызывая серьезные нарушения в работе мышц и трудности с дыханием.
Она развивается как у детей, так и у взрослых, вызывая серьезные нарушения в работе мышц и трудности с дыханием.
Причина заболевания кроется в мутации гена GAA, изменения в котором снижают активность фермента кислой альфа-глюкозидазы. Задача этого фермента — запускать переработку гликогена в клетках. Когда альфа-глюкозидазы не хватает, гликоген накапливается в разных органах и нарушает их работу. Оседая в скелетных мышцах, это вещество нарушает структуру мышечных волокон и лишает их способности полноценно сокращаться. Человек слабеет, теряет двигательные функции.
Болезнь впервые была описана в 1932 году голландским исследователем Иоганном Кассианусом Помпе, по имени которого и получила свое название. С тех пор ученые описали больше 360 генетических мутаций, запускающих патологический процесс накопления гликогена. Чем раньше он начинается, тем тяжелее протекает заболевание, поэтому принято выделять три основные формы болезни: младенческую (инфантильную), а также юношескую и взрослую, которые обычно объединяют в одну форму — болезнь Помпе с поздним дебютом.
Младенческая форма болезни Помпе
Самая тяжелая форма заболевания — младенческая. Ее симптомы появляются в первые месяцы жизни ребенка, затем болезнь быстро прогрессирует и часто приводит к летальному исходу еще в первый год жизни из-за сердечно-легочной недостаточности или инфекций дыхательных путей.
У 11-летнего Максима из Краснодарского края инфантильная форма болезни Помпе начала прогрессировать с третьего месяца жизни. Два года мальчик провел в реанимации, а с 2016 года находится на аппарате ИВЛ. Он не может самостоятельно сидеть, дышать и есть, но, несмотря на все трудности, разговаривает и даже учится на домашнем обучении. Современной ферментозаместительной терапией мальчика обеспечивает фонд «Круг добра».
«Наверное, я до сих пор ещё не приняла это заболевание. Мы все относимся к Максиму как здоровому полноценному ребёнку. Для нас он не инвалид, а просто особенный ребенок, — рассказала порталу „Помощь редким“ в прошлом году мама мальчика Валентина, — я не стесняюсь того, что он на аппарате.
Болезнь Помпе у взрослых
Заболевание, связанное с накоплением гликогена в организме, может проявиться и во взрослом возрасте. На ранних стадиях болезни взрослые люди обычно замечают:
- мышечную слабость (99%),
- трудности при беге (32%), при восхождении по лестнице (26%), при занятии спортом (23%),
- усталость и боль в мышцах (17%),
- трудности при ходьбе (16%), при поднятии с кресла (12%), при вставания из положения лежа (10%),
- дыхательные расстройства (1%).
Эта форма болезни, хоть и считается более «легкой», без необходимого лечения быстро прогрессирует. Международное исследование 2005 года показало, что около 50% больных через 10-15 лет после постановки диагноза нуждаются в инвалидных креслах и респираторной поддержке.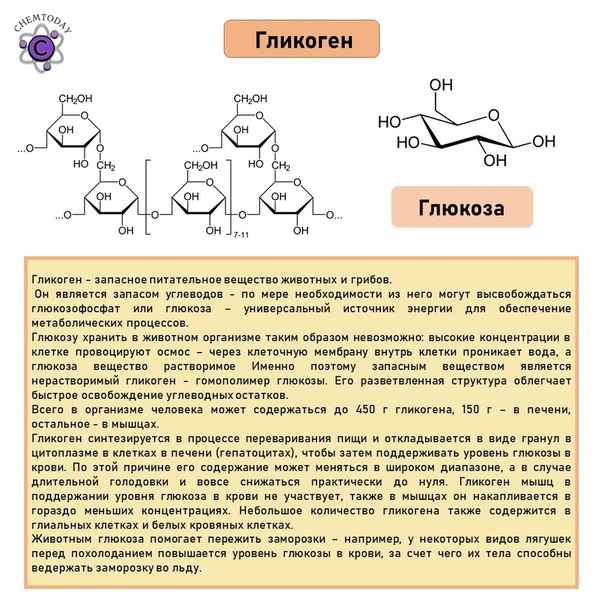
У Инны Становой, автора блога о миопатии и куратора Всероссийского общества орфанных заболеваний, болезнь Помпе диагностировали после 30 лет, когда появились серьезные двигательные проблемы: девушка не могла самостоятельно выйти из машины, встать со стула без помощи рук. На постановку диагноза ушло больше девяти месяцев. На тот момент в России уже был зарегистрировано лекарство для лечения болезни Помпе.
Пятый год, каждые две недели, Инна получает инфузии ферментозаместительным препаратом. «Теоретически препарат может только остановить прогрессирование болезни. Но у меня через полгода после начала терапии реально укрепились мышцы. Конечно, я ещё каждый день тренируюсь и правильно питаюсь, чтобы поддержать свой организм. В совокупности это даёт потрясающий результат», — говорит Инна.
Перспективы лечение болезни Помпе
До недавнего времени терапия болезни Помпе ограничивалась только симптоматическим лечением и профилактикой осложнений. Например, чтобы предотвратить хрупкость костей, которая развивается при болезни Помпе, пациентам назначались препараты кальция и диета, обогащенная кальцием и фосфором.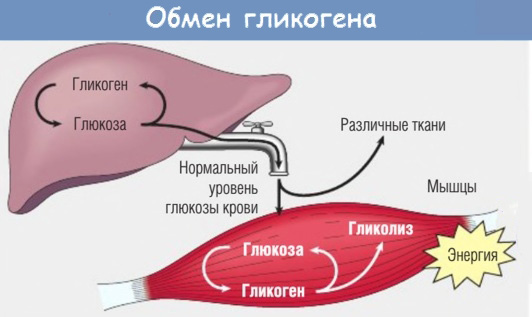 В наше время приоритетным способом лечения является ферментозаместительная терапия. Для этого используется препарат с активным веществом алглюкозидаза альфа.
В наше время приоритетным способом лечения является ферментозаместительная терапия. Для этого используется препарат с активным веществом алглюкозидаза альфа.
В феврале 2023 года также начались клинические исследования нового экспериментального перорального препарата для лечения болезни Помпе. Разработка принадлежит биотехнологической компании из Калифорнии Maze Therapeutics. Препарат под названием MZE001 подавляет активность гликогенсинтазы (GYS1) — фермента, который вырабатывает гликоген в мышечных клетках. Ожидается, что терапия снизит выработку гликогена, предотвратит его токсичное накопление и тем самым замедлит прогрессирование заболевания.
Больших успехов удалось достигнуть врачам в ранней диагностике и лечении младенческой формы заболевания. В 2022 году ученые описали первый случай успешного внутриутробного лечения болезни Помпе. 37-летней пациентке в первом триместре беременности пренатально с помощью биопсии ворсин хориона диагностировали патологию плода — болезнь Помпе. В качестве терапии ребенку вводили алглюкозидазу альфа (20 мг на килограмм расчетной массы плода) под ультразвуковым контролем через пупочную вену с 24-й до 34-й недели беременности.
В качестве терапии ребенку вводили алглюкозидазу альфа (20 мг на килограмм расчетной массы плода) под ультразвуковым контролем через пупочную вену с 24-й до 34-й недели беременности.
В ноябре 2022 года, на момент выхода научной публикации, девочке было 16 месяцев. Функция сердца, физическое и умственное развитие, а также уровни биомаркеров ребенка соответствовали возрастной норме.
По словам исследователей, ранняя диагностика и внутриутробное лечение болезни Помпе позволят избежать тяжелых повреждений внутренних органов младенца и являются началом новой главы в терапии плода.
Источники
- Мухамбетова Г.А., Усембаева Р.Б., Казанцев Е.О., Жуманазарова М.М. Болезнь Помпе – орфанное заболевание // Вестник КазНМУ. 2014. №2-1. URL: https://cyberleninka.ru/article/n/bolezn-pompe-orfannoe-zabolevanie (дата обращения: 11.04.2023).
- Семячкина А.Н., Сухоруков В.С., Букина Т.М., Яблонская М.И., Меркурьева Е.С., Харабадзе М.Н., Проскурина Е.
 А., Захарова Е.Ю., Брыдун А.В, Шаталов П.А, Новиков П.В. Болезнь накопления гликогена, тип II (болезнь Помпе) у детей // Российский вестник перинатологии и педиатрии. 2014. №4. URL: https://cyberleninka.ru/article/n/bolezn-nakopleniya-glikogena-tip-ii-bolezn-pompe-u-detey (дата обращения: 11.04.2023).
А., Захарова Е.Ю., Брыдун А.В, Шаталов П.А, Новиков П.В. Болезнь накопления гликогена, тип II (болезнь Помпе) у детей // Российский вестник перинатологии и педиатрии. 2014. №4. URL: https://cyberleninka.ru/article/n/bolezn-nakopleniya-glikogena-tip-ii-bolezn-pompe-u-detey (дата обращения: 11.04.2023).
теги
Болезнь Помпе
Болезнь накопления гликогена IV типа: MedlinePlus Genetics
Описание
Болезнь накопления гликогена IV типа (GSD IV) — это наследственное заболевание, вызванное накоплением сложного сахара, называемого гликогеном, в клетках организма. Накопленный гликоген является структурно аномальным и нарушает функцию некоторых органов и тканей, особенно печени и мышц. Существует пять типов GSD IV, которые различаются по степени тяжести, признакам и симптомам.
Перинатальный нервно-мышечный тип со смертельным исходом является наиболее тяжелой формой GSD IV, симптомы которой развиваются до рождения.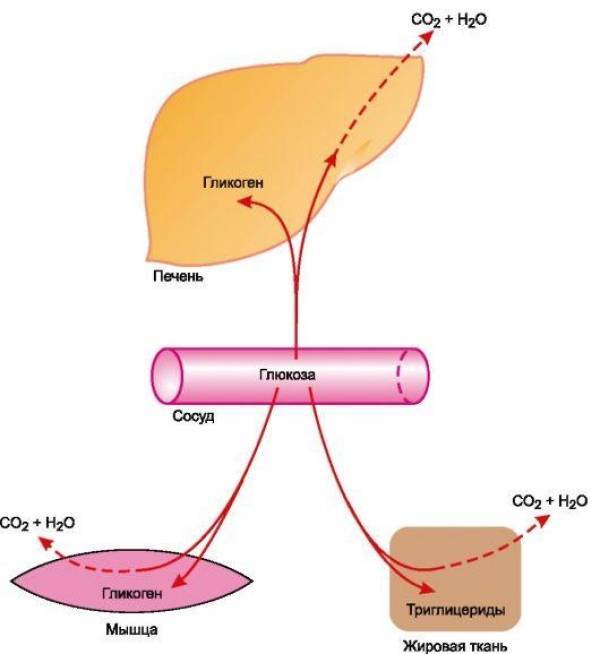 Избыток жидкости может накапливаться вокруг плода (многоводие) и в теле плода. Пораженные плоды имеют состояние, называемое последовательностью деформации акинезии плода, которое вызывает снижение подвижности плода и может привести к тугоподвижности суставов (артрогрипоз) после рождения. Младенцы с фатальным перинатальным нервно-мышечным типом GSD IV имеют очень низкий мышечный тонус (тяжелая гипотония) и атрофию мышц. Эти младенцы обычно не доживают до периода новорожденности из-за ослабления сердечной и дыхательной мускулатуры.
Избыток жидкости может накапливаться вокруг плода (многоводие) и в теле плода. Пораженные плоды имеют состояние, называемое последовательностью деформации акинезии плода, которое вызывает снижение подвижности плода и может привести к тугоподвижности суставов (артрогрипоз) после рождения. Младенцы с фатальным перинатальным нервно-мышечным типом GSD IV имеют очень низкий мышечный тонус (тяжелая гипотония) и атрофию мышц. Эти младенцы обычно не доживают до периода новорожденности из-за ослабления сердечной и дыхательной мускулатуры.
Врожденный мышечный тип GSD IV обычно не проявляется до рождения, но развивается в раннем младенчестве. У пораженных младенцев наблюдается тяжелая гипотония, которая влияет на мышцы, необходимые для дыхания. У этих детей часто наблюдается дилатационная кардиомиопатия, которая увеличивает и ослабляет сердечную (сердечную) мышцу, не позволяя сердцу эффективно перекачивать кровь. Младенцы с врожденным мышечным типом GSD IV обычно выживают всего несколько месяцев.
Прогрессирующий печеночный тип является наиболее распространенной формой GSD IV. В течение первых месяцев жизни больные дети с трудом набирают вес и растут с ожидаемой скоростью (отставание в развитии), у них увеличивается печень (гепатомегалия). У детей с этим типом развивается необратимая форма заболевания печени, называемая циррозом. Также могут возникать высокое кровяное давление в венах, которые снабжают кровью печень (портальная гипертензия), и аномальное накопление жидкости в брюшной полости (асцит). К 1 или 2 годам у больных детей развивается гипотония. Дети с прогрессирующим печеночным типом ЖКБ IV часто умирают от печеночной недостаточности в раннем детстве.
Непрогрессирующий печеночный тип GSD IV имеет многие из тех же признаков, что и прогрессирующий печеночный тип, но заболевание печени не такое тяжелое. При непрогрессирующем печеночном типе гепатомегалия и заболевание печени обычно проявляются в раннем детстве, но у больных обычно не развивается цирроз. Люди с этим типом расстройства также могут иметь гипотонию и мышечную слабость (миопатию).
Детский нервно-мышечный тип GSD IV развивается в позднем детстве и характеризуется миопатией и дилатационной кардиомиопатией. Тяжесть этого типа GSD IV сильно различается; у некоторых людей наблюдается лишь легкая мышечная слабость, в то время как у других развивается тяжелая кардиомиопатия, и они умирают в раннем взрослом возрасте.
Частота
GSD IV, по оценкам, возникает у 1 из 600 000–800 000 человек во всем мире. На тип IV приходится примерно 3 процента всех случаев болезни накопления гликогена.
Причины
Мутации в гене GBE1 вызывают GSD IV. Ген GBE1 предоставляет инструкции по получению фермента разветвления гликогена. Этот фермент участвует в производстве гликогена, который является основным источником запасенной энергии в организме. Мутации гена GBE1 , вызывающие GSD IV, приводят к недостатку (дефициту) фермента ветвления гликогена.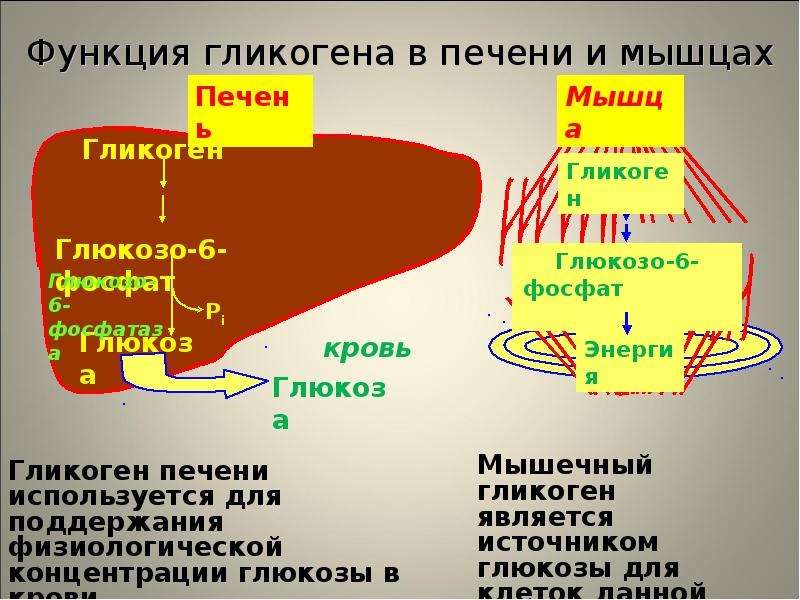 В результате гликоген не образуется должным образом. Аномальные молекулы гликогена, называемые полиглюкозановыми тельцами, накапливаются в клетках, что приводит к повреждению и гибели клеток. Полиглюкозановые тельца накапливаются в клетках по всему телу, но клетки печени и мышечные клетки наиболее сильно поражаются при GSD IV. Накопление гликогена в печени приводит к гепатомегалии и нарушению функции печени. Неспособность мышечных клеток расщеплять гликоген для получения энергии приводит к мышечной слабости и истощению.
В результате гликоген не образуется должным образом. Аномальные молекулы гликогена, называемые полиглюкозановыми тельцами, накапливаются в клетках, что приводит к повреждению и гибели клеток. Полиглюкозановые тельца накапливаются в клетках по всему телу, но клетки печени и мышечные клетки наиболее сильно поражаются при GSD IV. Накопление гликогена в печени приводит к гепатомегалии и нарушению функции печени. Неспособность мышечных клеток расщеплять гликоген для получения энергии приводит к мышечной слабости и истощению.
Как правило, тяжесть заболевания связана с количеством вырабатываемого функционального фермента ветвления гликогена. Лица с фатальным перинатальным нервно-мышечным типом, как правило, производят менее 5 процентов пригодных для использования ферментов, в то время как люди с нервно-мышечным типом детского возраста могут иметь около 20 процентов функции фермента. Другие типы GSD IV обычно связаны с от 5 до 20 процентов рабочего фермента. Однако эти оценки различаются для разных типов.
Наследование
Это состояние наследуется по аутосомно-рецессивному типу, что означает, что обе копии гена в каждой клетке имеют мутации. Каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутировавшего гена, но обычно у них нет признаков и симптомов заболевания.
Другие названия для этого состояния
- Амилопектиноз
- Болезнь Андерсена
- Андерсеновский гликогеноз
- Болезнь Андерсена
- Дефицит ветвления
- Дефицит фермента ветвления
- Дефицит фермента ветвления гликогена
- Болезнь накопления гликогена IV
- Болезнь накопления гликогена 4 типа
- Гликогеноз 4
- G ликогеноз, тип IV
- GSD IV
- GSD тип IV
- GSD4
- Гликогеноз IV типа
Дополнительная информация и ресурсы
Информация о генетическом тестировании
- Реестр генетического тестирования: Болезнь накопления гликогена, тип IV
Информационный центр генетических и редких заболеваний
- Болезнь накопления гликогена 4 типа
Ресурсы поддержки и защиты интересов пациентов
- Информационный поиск по болезням
- Национальная организация редких заболеваний (NORD)
Научные исследования от ClinicalTrials.
 gov
gov- ClinicalTrials.gov
Каталог генов и болезней от OMIM
- БОЛЕЗНЬ НАКОПЛЕНИЯ ГЛИКОГЕНА IV
Научные статьи в PubMed
- PubMed
Каталожные номера
- Ассерето С., ван Диггелен О.П., Диого Л., Морава Э., Кассандрини Д., Каррейра И., де Boode WP, Dilling J, Garcia P, Henriques M, Rebelo O, ter Laak H, Minetti C, Бруно К. Нулевые мутации и летальная врожденная форма болезни накопления гликогена тип IV. Biochem Biophys Res Commun. 21 сентября 2007 г .; 361 (2): 445–50. дои: 10.1016/j.bbrc.2007.07.074. Epub 2007, 24 июля. Цитирование на PubMed
- Бруно С., Кассандрини Д., Ассерето С., Акман Х.О., Минетти С., Ди Мауро С. Нервно-мышечные формы недостаточности фермента ветвления гликогена. Акта Миол. 2007 г. 26 июля (1): 75-8. Цитирование в PubMed или бесплатная статья в PubMed Central
- Бруно С., ван Диггелен О.П.
 , Кассандрини Д., Гимпелев М., Джуффре Б., Донати М.А.,
Интровини П., Алегрия А., Ассерето С., Моранди Л., Мора М., Тоноли Э., Масчелли С.,
Траверсо М., Паскини Э., Бадо М., Вилариньо Л., ван Ноорт Г., Моска Ф., ДиМауро С.,
Zara F, Minetti C. Клиническая и генетическая гетерогенность ветвящегося фермента
дефицит (гликогеноз IV типа). Неврология. 2004 г., 28 сентября; 63 (6): 1053-8. дои:
10.1212/01.внл.0000138429.11433.0д. Цитата в PubMed
, Кассандрини Д., Гимпелев М., Джуффре Б., Донати М.А.,
Интровини П., Алегрия А., Ассерето С., Моранди Л., Мора М., Тоноли Э., Масчелли С.,
Траверсо М., Паскини Э., Бадо М., Вилариньо Л., ван Ноорт Г., Моска Ф., ДиМауро С.,
Zara F, Minetti C. Клиническая и генетическая гетерогенность ветвящегося фермента
дефицит (гликогеноз IV типа). Неврология. 2004 г., 28 сентября; 63 (6): 1053-8. дои:
10.1212/01.внл.0000138429.11433.0д. Цитата в PubMed - Берроу Т.А., Хопкин Р.Дж., Бове К.Е., Майлз Л., Вонг Б.Л., Чоудхари А., Бали Д., Ли СК, Чен Ю.Т. Нелетальная врожденная гипотония вследствие болезни накопления гликогена IV типа. Am J Med Genet A. 15 апреля 2006 г.; 140 (8): 878-82. doi: 10.1002/ajmg.a.31166. Цитата в PubMed
- Фернандес К., Халберт К., Де Паула А.М., Лакроз В., Фруассар Р., Фигарелла-Бранже
Д., Шаброль Б., Пеллиссье Ж.Ф. Нелетальный неонатальный нервно-мышечный вариант
гликогеноз IV типа с новыми мутациями GBE1. Мышечный нерв. 2010
Фев; 41 (2): 269-71.
 doi: 10.1002/mus.21499. Цитата в PubMed
doi: 10.1002/mus.21499. Цитата в PubMed - Magoulas PL, El-Hattab AW, Roy A, Bali DS, Finegold MJ, Craigen WJ. Диффузный поражение ретикулоэндотелиальной системы при болезни накопления гликогена IV типа с новая мутация GBE1: клинический случай и обзор. Хум Патол. 2012 июнь;43(6):943-51. doi: 10.1016/j.humpath.2011.10.001. Epub 2012 Feb 2. Цитата на PubMed
- Тай С.К., Акман Х.О., Чанг В.К., Пайк М.Г., Мунтони Ф., Хейс А.П., Шанске С., Вальберг SJ, Mickelson JR, Tanji K, DiMauro S. Фатальное младенческое нервно-мышечное проявление болезнь накопления гликогена IV типа. Нервно-мышечное расстройство. 2004 г., 14 апреля (4): 253–60. doi: 10.1016/j.nmd.2003.12.006. Цитата на PubMed
Болезнь Помпе: MedlinePlus Genetics
Описание
Болезнь Помпе — это наследственное заболевание, вызванное накоплением в клетках организма сложного сахара, называемого гликогеном. Накопление гликогена в некоторых органах и тканях, особенно в мышцах, нарушает их способность нормально функционировать.
Исследователи описали три типа болезни Помпе, которые различаются по степени тяжести и возрасту, в котором они появляются. Эти типы известны как классическое инфантильное начало, неклассическое инфантильное начало и позднее начало.
Классическая форма детской болезни Помпе начинается в течение нескольких месяцев после рождения. Младенцы с этим расстройством обычно испытывают мышечную слабость (миопатия), плохой мышечный тонус (гипотония), увеличение печени (гепатомегалия) и пороки сердца. Больные младенцы также могут не набирать вес и расти с ожидаемой скоростью (отставание в развитии) и иметь проблемы с дыханием. При отсутствии лечения эта форма болезни Помпе приводит к смерти от сердечной недостаточности на первом году жизни.
Неклассическая форма болезни Помпе с дебютом в младенческом возрасте обычно проявляется в возрасте 1 года. Она характеризуется задержкой двигательных навыков (таких как переворачивание и сидение) и прогрессирующей мышечной слабостью. Сердце может быть аномально большим (кардиомегалия), но у больных обычно не возникает сердечной недостаточности. Мышечная слабость при этом расстройстве приводит к серьезным проблемам с дыханием, и большинство детей с неклассической болезнью Помпе с инфантильным началом доживают только до раннего детства.
Мышечная слабость при этом расстройстве приводит к серьезным проблемам с дыханием, и большинство детей с неклассической болезнью Помпе с инфантильным началом доживают только до раннего детства.
Болезнь Помпе с поздним началом может проявиться только в детстве, подростковом или взрослом возрасте. Болезнь Помпе с поздним началом обычно протекает легче, чем формы этого расстройства с инфантильным началом, и с меньшей вероятностью поражает сердце. Большинство людей с поздним началом болезни Помпе испытывают прогрессирующую мышечную слабость, особенно в ногах и туловище, включая мышцы, контролирующие дыхание. По мере прогрессирования расстройства проблемы с дыханием могут привести к дыхательной недостаточности.
Частота
Болезнь Помпе поражает примерно 1 из 40 000 человек в Соединенных Штатах. Заболеваемость этим расстройством варьируется среди различных этнических групп.
Причины
Мутации в гене GAA вызывают болезнь Помпе. Ген GAA предоставляет инструкции для производства фермента, называемого кислой альфа-глюкозидазой (также известной как кислая мальтаза). Этот фермент активен в лизосомах, которые представляют собой структуры, служащие центрами рециркуляции внутри клеток. Фермент обычно расщепляет гликоген на более простой сахар, называемый глюкозой, который является основным источником энергии для большинства клеток.
Этот фермент активен в лизосомах, которые представляют собой структуры, служащие центрами рециркуляции внутри клеток. Фермент обычно расщепляет гликоген на более простой сахар, называемый глюкозой, который является основным источником энергии для большинства клеток.
Мутации в гене GAA препятствуют эффективному расщеплению гликогена кислой альфа-глюкозидазой, что позволяет этому сахару накапливаться до токсического уровня в лизосомах. Это накопление повреждает органы и ткани по всему телу, особенно мышцы, что приводит к прогрессирующим признакам и симптомам болезни Помпе.
Наследование
Это состояние наследуется по аутосомно-рецессивному типу, что означает, что обе копии гена в каждой клетке имеют мутации. Каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутировавшего гена, но обычно у них нет признаков и симптомов заболевания.
Другие названия для этого состояния
- Дефицит кислой мальтазы
- Болезнь, вызванная дефицитом кислой мальтазы
- Дефицит альфа-1,4-глюкозидазы
- AMD
- Дефицит альфа-глюкозидазы 900 40
- Дефицит ГАА
- Болезнь накопления гликогена II типа
- Гликогеноз II типа
- GSD II
- GSD2
- Болезнь Помпе
Дополнительная информация и ресурсы
Информация о генетическом тестировании
- Реестр генетического тестирования: Болезнь накопления гликогена II типа, инфантильная
- Реестр генетического тестирования: Болезнь накопления гликогена, тип II
Информационный центр генетических и редких заболеваний
- Болезнь накопления гликогена 2 типа
Ресурсы поддержки и защиты интересов пациентов
- Информационный поиск по болезням
- Национальная организация редких заболеваний (NORD)
Научные исследования от ClinicalTrials.
 gov
gov- ClinicalTrials.gov
Каталог генов и болезней от OMIM
- БОЛЕЗНЬ НАКОПЛЕНИЯ ГЛИКОГЕНА II
Научные статьи в PubMed
- PubMed
Ссылки
- Bembi B, Cerini E, Danesino C, Donati MA, Gasperini S, Morandi L, Musumeci O, Паренти Г., Равалья С., Сеидита Ф., Тоскано А., Вианелло А. Диагностика гликогеноз II типа. Неврология. 2 декабря 2008 г .; 71 (23 Дополнение 2): S4-11. дои: 10.1212/WNL.0b013e31818da91е. Цитата в PubMed
- Чиен Ю.Х., Ли Н.К., Терберг Б.Л., Чанг С.К., Чжан С.К., Койцер Дж., Хуан А.С., Ву М.Х., Хуан П.Х., Цай Ф.Дж., Чен Ю.Т., Хву В.Л. Болезнь Помпе у детей раннего возраста: улучшение прогноз с помощью скрининга новорожденных и раннего лечения. Педиатрия. 2009 г. Декабрь; 124 (6): e1116-25. doi: 10.1542/пед.2008-3667. Цитата в PubMed
- Фукуда Т., Робертс А., Плотц П.Х., Рабен Н. Дефицит кислой альфа-глюкозидазы
(болезнь Помпе).
 Curr Neurol Neurosci Rep. 2007 Jan; 7(1):71-7. дои:
10.1007/с11910-007-0024-4. Цитата в PubMed
Curr Neurol Neurosci Rep. 2007 Jan; 7(1):71-7. дои:
10.1007/с11910-007-0024-4. Цитата в PubMed - Кишнани П.С., Хву В.Л., Мандель Х., Николино М., Йонг Ф., Корзо Д.; Инфантильное начало Группа изучения естественной истории болезни Помпе. Ретроспективный, многонациональный, многоцентровое исследование естественного течения болезни Помпе с инфантильным началом. Дж Педиатр. 2006 г., май; 148(5):671-676. doi: 10.1016/j.jpeds.2005.11.033. Цитата в PubMed
- Кишнани П.С., Штайнер Р.Д., Бали Д., Бергер К., Бирн Б.Дж., Кейс Л.Е., Кроули Дж.Ф.,
Даунс С., Хауэлл Р. Р., Кравиц Р. М., Макки Дж., Марсден Д., Мартинс А. М., Миллингтон Д. С.,
Николино М., О’Грэйди Г., Паттерсон М.С., Рапопорт Д.М., Слоним А., Спенсер К.Т., Тиффт К.Дж.,
Уотсон МС. Руководство по диагностике и лечению болезни Помпе. Генет Мед. 2006 г.
Май; 8 (5): 267-88. дои: 10.1097/01.гим.0000218152.87434.f3. Аннотация недоступна.
Опечатка в: Genet Med. 2006 г., июнь; 8 (6): 382. Рабочая группа ACMG по управлению Pompe
Болезнь [удалено]; Кейс, Лаура [исправлено на Кейс, Лора Э.