Обмен отдельных аминокислот — презентация онлайн
ОБМЕН ОТДЕЛЬНЫХ
АМИНОКИСЛОТ
2. Метаболизм глицина, серина, треонина.
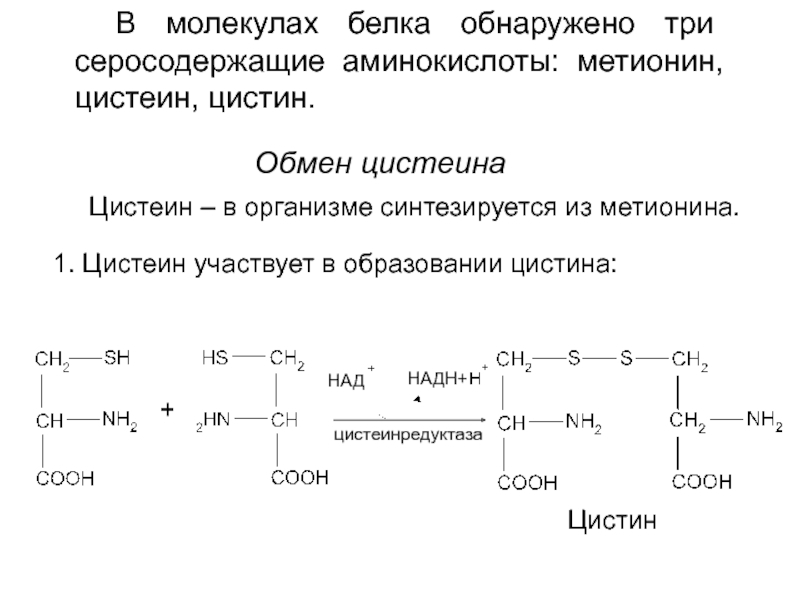
4. Обмен серосодержащих аминокислот. Цистеин.
5. Взаимосвязь обмена серина, глицина, метионина и цистеина:
6. Трансметилирование
8. Синтез креатина
Протекает в 2х органах : почках и печениКреатин-Ф играет большую роль особенно для мышц , поскольку
поддерживает соотношение АТФ к АДФ в мышцах.
10. Обмен одноуглеродных фрагментов.
12. Недостаточность фолиевой кислоты.
Недостаточность фолиевой кислоты у человекавозникает редко. Гиповитаминоз фолиевой кислоты
приводит к нарушению обмена одноуглеродных
фрагментов.
Проявления недостаточности фолиевой кислоты:
-Первое проявление дефицита фолиевой кислоты –
мегалобластная анемия. Она характеризуется
уменьшением количества эритроцитов, снижением
содержания в них гемоглобина, что вызывает
увеличение размеров эритроцитов.

— Лейкопения и тромбоцитопения.
— Подавление активности иммунных реакций.
— Снижение фагоцитарной активности гранулоцитов.
— Ослабление резистентности организма к возбудителям
инфекции (преимущественно вирусной природы).
Фолиевая кислота
15. Обмен фенилаланина и тирозина.
17. Фенилкетонурия.
Классическая ФКУ — наследственное заболевание, связанное смутациями в гене фенилаланингидроксилазы, которые
приводят к снижению активности фермента или полной его
инактивации.
Наиболее тяжёлые проявления ФКУ — нарушение умственного и
физического развития, судорожный синдром, нарушение
пигментации. При отсутствии лечения больные не доживают до 30
лет.
Тяжёлые проявления ФКУ связаны с токсическим действием на
клетки мозга высоких концентраций фенилаланина,
фенилпирувата, фениллактата. Большие концентрации
фенилаланина ограничивают транспорт тирозина и триптофана
через гематоэнцефаличеекий барьер и тормозят синтез нейромедиаторов (дофамина, норадреналина, серотонина).
Вариантная ФКУ (коферментзависимая гиперфенилаланинемия) следствие мутаций в генах, контролирующих метаболизм Н4БП.
Заболевание характеризуется тяжёлыми неврологическими
нарушениями и ранней смертью («злокачественная» ФКУ).
Симптомы фенилкетонурии:
Ребенок умственно отсталый, возбудим, своеобразная
походка, осанка и поза при сидении, конечности находятся в
необычном положении, стереотипность движений,
сухожильные рефлексы повышены, возможны судороги,
микроцефалия, гипопигментация, экзема,
гипопигментированность волос, катаракта, своеобразный
запах тела.
Лечение фенилкетонурии:
Больной должен соблюдать диету — продукты не должны
содержать фенилаланин. Исключены мясные блюда, блюда
из птицы, а также рыбные, грибные и молочные. Белок
компенсируется специальными смесями аминокислот с
малым содержанием фенилаланина.
20. Тирозинемии.
Тирозинемия типа I (тирозиноз).Причиной заболевания является, вероятно, дефект фермента
фумарилацетоацетатгидролазы, катализирующего расщепление
фумарилацетоа-цетата на фумарат и ацетоацетат.
 Накапливающиеся
Накапливающиесяметаболиты снижают активность некоторых ферментов и
транспортных систем аминокислот. Патофизиология этого
нарушения достаточно сложна. Острая форма тирозиноза
характерна для новорождённых. Клинические проявления — диарея,
рвота, задержка в развитии. Без лечения дети погибают в возрасте 68 мес из-за развивающейся недостаточности печени.Хроническая
форма характеризуется сходными, но менее выраженными
симптомами. Гибель наступает в возрасте 10 лет. Содержание
тирозина в крови у больных в несколько раз превышает норму. Для
лечения используют диету с пониженным содержанием тирозина и
фенилаланина.
Тирозинемия типа II (синдром Рихнера-Ханхорта).
Причина — дефект фермента
тирозинаминотрансферазы. Концентрация тирозина
в крови больных повышена. Для заболевания
характерны поражения глаз и кожи, умеренная
умственная отсталость, нарушение координации
движений.
Тирозинемия новорождённых (кратковременная).
Заболевание возникает в результате снижения
активности фермента
гидроксифенилпируватдиоксигеназы,
превращающего гидроксифенилпируват в
гомогентизиновую кислоту.
 В результате в крови
В результате в кровибольных повышается концентрация
гидроксифенилацетата, тирозина и фенил-аланина.
При лечении назначают бедную белком диету и
витамин С.
Поражение кожи при тирозинемии.
23. Алкаптонурия («чёрная моча»)
Алкаптонурия («чёрная моча»)Причина заболевания — дефект диоксигеназы
гомогентизиновой кислоты. Для этой болезни
характерно выделение с мочой большого
количества гомогентизиновой кислоты, которая,
окисляясь кислородом воздуха, образует тёмные
пигменты алкаптоны.
Клиническими проявлениями болезни, кроме
потемнения мочи на воздухе, являются
пигментация соединительной ткани (охроноз) и
артрит.
26. Альбинизм.
Причина метаболического нарушения —врождённый дефект тирозиназы. Этот фермент
катализирует превращение тирозина в ДОФА в
меланоцитах. В результате дефекта тирозиназы
нарушается синтез пигментов меланинов.
Клиническое проявление альбинизма
(от лат. albus — белый) — отсутствие пигментации
кожи и волос.
 У больных часто снижена острота
У больных часто снижена остротазрения, возникает светобоязнь. Длительное
пребывание таких больных под открытым солнцем
приводит к раку кожи.
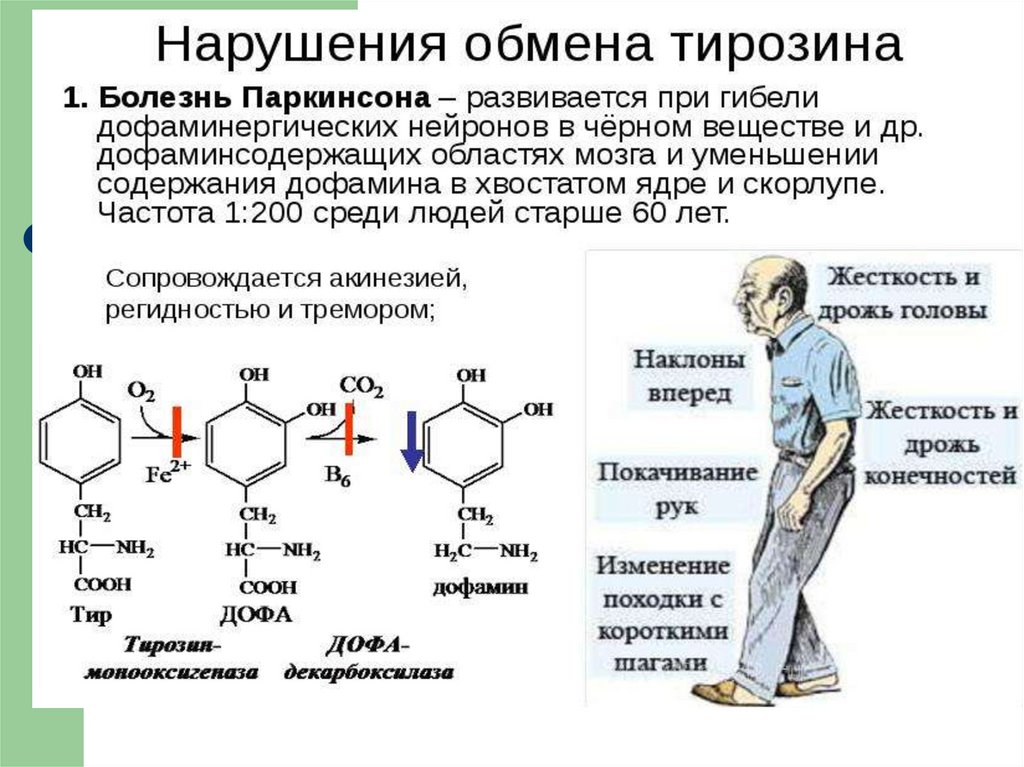
28. Болезнь Паркинсона.
Заболевание развивается при недостаточности дофамина вчёрной субстанции мозга. При этой патологии снижена
активность тирозингидроксилазы, ДОФАдекарбоксилазы. Заболевание сопровождается тремя
основными симптомами:
акинезия (скованность движений),
ригидность (напряжение мышц),
тремор (непроизвольное дрожание).
Для лечения паркинсонизма предлагаются следующие
принципы:
Заместительная терапия препаратами-предшественниками
дофамина (производными ДОФА) — леводопа, мадопар,
наком и др.
Подавление инактивации дофамина ингибиторами МАО
(депренил, ниаламид, пиразидол и др.).
31. Болезнь мочи кленового сиропа.
БМКС вызвана дефицитом комплекса дегидрогеназыальфа-кетокислот с разветвленной цепью, вследствие
чего в крови и моче происходит накопление аминокислот с
разветвленной углеродной цепью (лейцина, изолейцина и
валина) и токсичных продуктов их метаболизма.

Заболевание характеризуется наличием сладкого запаха
мочи у маленьких детей (запах аналогичный запаху
кленового сиропа). При рождении у детей нет никаких
видимых признаков заболевания. Однако, если
расстройство не лечить, то у больных возникают серьезные
повреждения головного мозга, которые могут привести к
смерти пораженного ребенка.
Лейциноз или болезнь мочи кленового сиропа
33. Болезнь Вильсона-Коновалова.
— врождённое нарушение метаболизма меди,приводящее к тяжелейшим наследственным болезням
центральной нервной системы и внутренних органов.
Нарушение метаболизма выражается в нарушении синтеза и
снижении в крови концентрации церулоплазмина.
Церулоплазмин участвует в процессе выведения меди из
организма. В печени формируется крупноузловой или
смешанный цирроз. В почках в первую очередь страдают
проксимальные канальцы. В головном мозге поражаются в
большей степени базальные ганглии, зубчатое ядро
мозжечка и черная субстанция.
 Отложение меди в
Отложение меди вдесцеметовой мембране глаза приводит к формированию
кольца Кайзера-Флейшера.
Типичным симптомом болезни является кольцо Кайзера-Флейшера
— отложение по периферии роговой оболочки содержащего медь
зеленовато-бурого пигмента; оно более выражено при поздних формах
заболевания. Иногда отмечается желтовато-коричневая пигментация
кожи туловища и лица. Часты геморрагические явления (кровоточивость
дёсен, носовые кровотечения, положительная проба жгута), мраморность
кожи, акроцианоз. Капилляроскопия обнаруживает атонию капилляров и
застойность кровотока. Отмечаются суставные боли, профузные поты,
остеопороз, ломкость костей.
36. Обмен триптофана
Особенности обмена отдельных аминокислот — презентация онлайн
Похожие презентации:
Обмен белков: Индивидуальные пути обмена аминокислот
Специфические пути обмена отдельных аминокислот. Патология. (Лекция 12)
Пути обмена отдельных аминокислот
Пути обмена отдельных аминокислот
Обмен белков и аминокислот.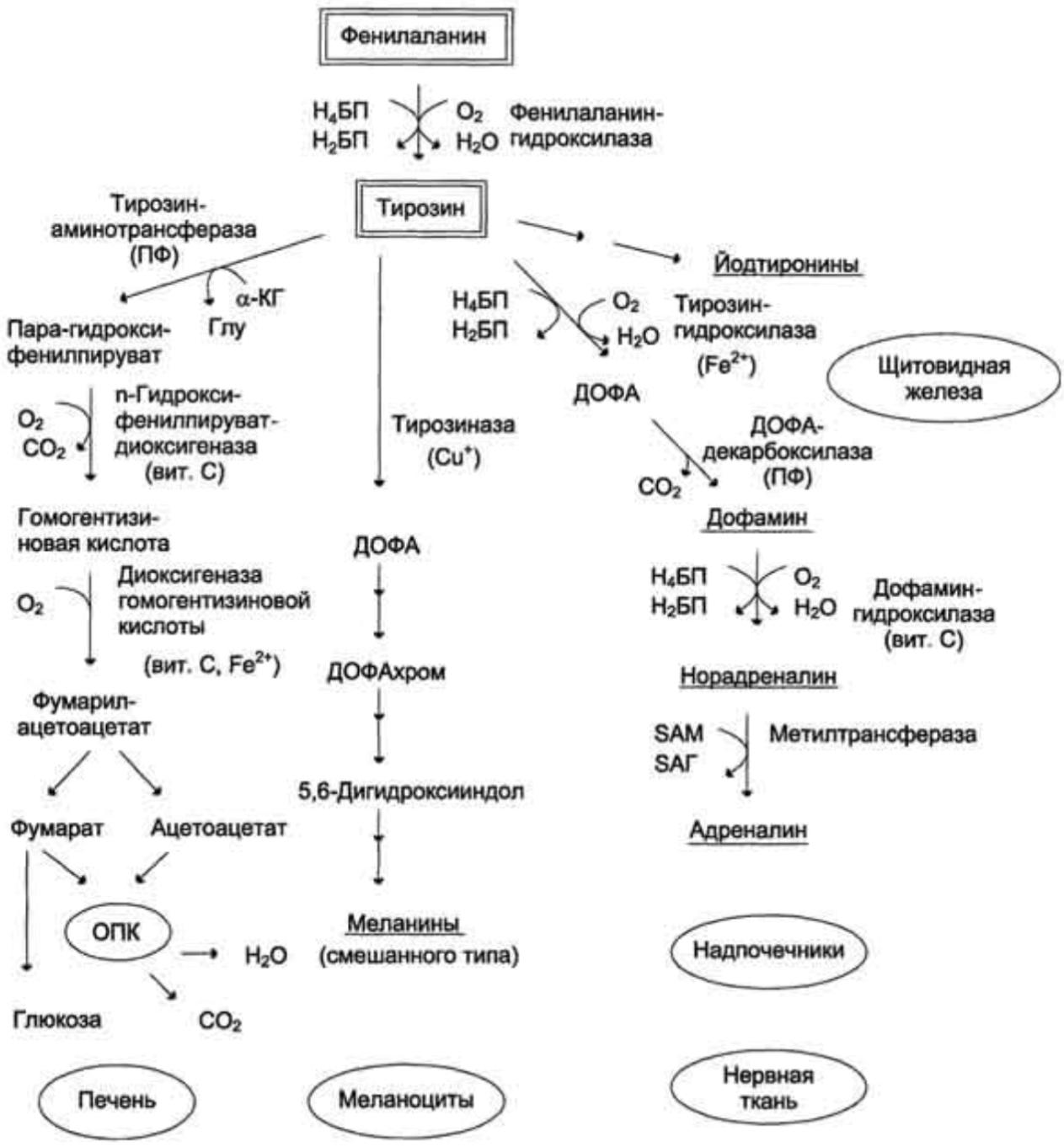 Азотистый баланс. (Лекция 14)
Азотистый баланс. (Лекция 14)
Метаболизм аминокислот (простых белков)
Индивидуальные пути обмена аминокислот. Часть 2. Лекция №14
Обмен серосодержащих аминокислот
Обезвреживание аммиака. Биосинтез мочевины
Обмен и функции аминокислот
Особенности
обмена
отдельных
аминокислот
Функции цистеина — участие в фолдинге
белков за счет способности тиогруппы
цистеина образовывать дисульфидные
связи.
При этом 2 остатка цистеина формируют
молекулу цистина.
Эта окислительная реакция протекает либо
неферментативно, либо с участием фермента
является NAD+
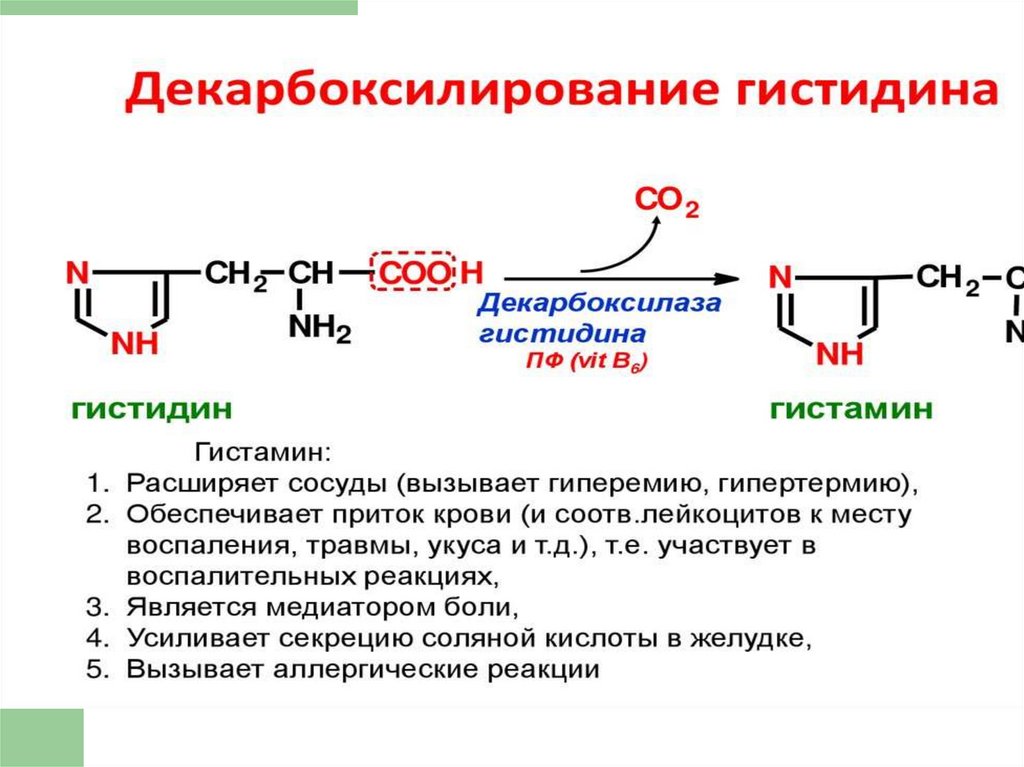
Нарушения обмена ЦИСТЕИНА
Образование гомоцистина при
нарушении использования гомоцистеина
Гомоцистин накапливается в крови и в тканях,
выделяется с мочой, вызывая гомоцистинурию.
Причины — гиповитаминоз фолиевой кислоты, а
также витаминов В6 и В12.
Дисульфидные связи стабилизируют
пространственную структуру полипептидной
цепи или связывают между собой 2 цепи
(например:А и В-цепи в молекуле инсулина).

Очень многие белки и ферменты содержат в
активном центре SH-группы, участвующие в
катализе. При их окислении ферментативная
активность падает.
Восстановление SH-групп часто происходит с
использованием глутатиона — трипептида,
содержащего гамма-глутаминовую кислоту,
цистеин и глицин. Глутатион имеет 2 формы:
восстановленную (Г-SH) и окисленную (Г-S-S-Г) и
является активным антиоксидантом.
5. Участие глутатиона в восстановлении цистина
-Осуществлениеантиоксидантной
функции
-Формирование
надвторичной
структур
-Участие в фолдинге
белка
СИНТЕЗ ТАУРИНА — важный путь использования
цистеина, который осуществляется за счет
декарбоксилирования производных цистеина цистеиновой и цистеинсульфиновой кислот:
ФУНКЦИИ ТАУРИНА синтез желчных кислот в печени
антиоксидантная защита
ОБЩАЯ СХЕМА ФУНКЦИЙ ЦИСТЕИНА
Белки
Глутатион
ЦИСТЕИН
Таурин
HS-КоА
Пируват ОПК Глюкоза
Сульфаты Моча
МЕТИОНИН — незаменимая АМК,
однако она может регенерироваться
из гомоцистеина.
Следовательно,
незаменим
именно
гомоцистеин,
но
единственным
его
источником в организме является метионин.
В пище гомоцистеина крайне мало, поэтому
потребности человека в гомоцистеине и
метионине
обеспечиваются
только
метионином пищи.
Общая схема метаболизма метионина
1
3
2
1-реакции трансметилирования, 2-синтез цистеина, 3-регенерация
метионина.
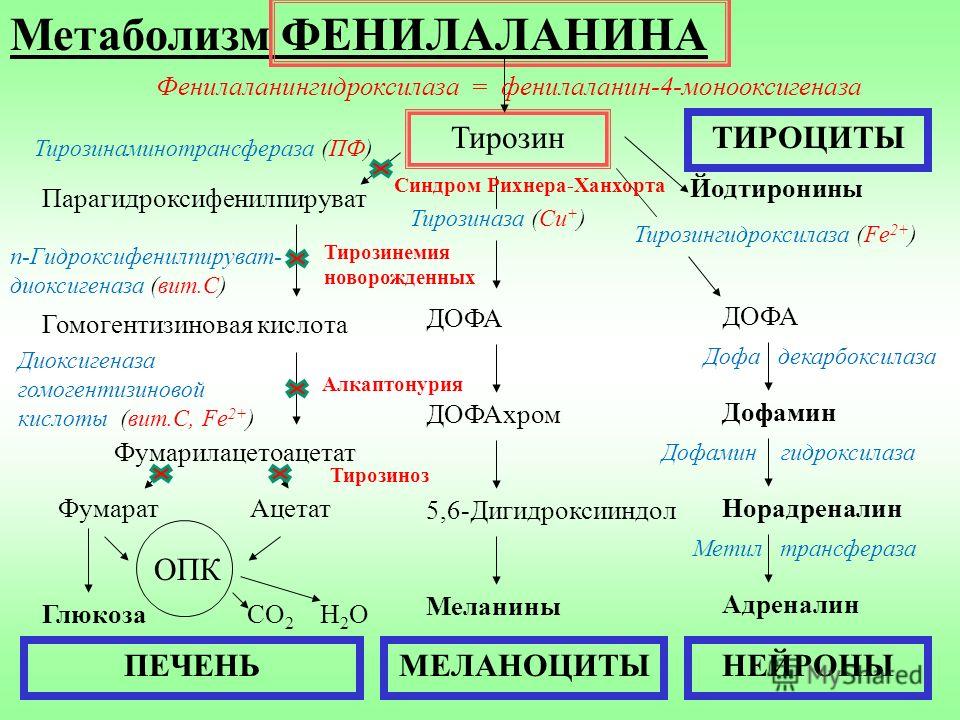
Метаболизм ФЕНИЛАЛАНИНА
2 основных пути: включение в белки
и превращение в тирозин
Тирозин — заменимая АМК, и превращение в нее
фенилалаланина путем гидроксилирования
необходимо только для удаления избытка
фенилаланина.
Реакция катализируется специфической
монооксигеназой — фенилаланингидроксилазой
(коферменты — тетрагидробиоптерин Н4БП и Fe2+)
.
Метаболизм ФЕНИЛАЛАНИНА
Фенилаланин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
гидроксилаза
Тирозин
Йодтиронины
Тирозиназа (Сu+)
Гидроксифенилпируватдиоксигеназа (вит.
 С)
С)Гомогентизиновая кислота
ДОФА
ДОФА
Ацетат
Дофамин
5,6-Дигидроксииндол
СО2
ПЕЧЕНЬ
Н2О
Меланины
МЕЛАНОЦИТЫ
гидроксилаза
Норадреналин
Метил
ОПК
декарбоксилаза
Дофамин
ДОФАхром
Фумарилацетоацетат
Глюкоза
Тирозингидроксилаза (Fe2+)
Дофа
Диоксигеназа
гомогентизиновой
кислоты (вит.С, Fe2+)
Фумарат
ТИРОЦИТЫ
трансфераза
Адреналин
НЕЙРОНЫ
12. Реакция превращения фенилаланина в тирозин
Обмен ФЕНИЛАЛАНИНА и ТИРОЗИНАсвязан со значительным количеством реакций
гидроксилирования, катализируемых оксигеназами
(гидроксилазами), использующими молекулу О2 и
коферменты — донор водорода (чаще Р4БП), Fe2+, Сu+,
гем, витамин С.
Оксигеназы делят на 2 группы:
1. Монооксигеназы (МАОА и МАОВ) — один атом О2
присоединяется к продукту реакции, другой используется
для образования Н2О.
2. Диоксигеназы — оба атома О2 используются для
образования продукта реакции.
 Диоксигеназы
Диоксигеназыкатализируют все процессы расщепления ароматических
колец в биологических системах.
При любых нарушениях
превращения его в
тирозин развивается
фенилкетонурия.
Схема превращение фенилаланина
при фенилкетонурии
В патогенезе фенилкетонурии имеют значение многие
обстоятельства, в частности:
1) значительное накопление в тканях и жидкостях больного организма
фенилаланина и его производных (фенилпировиноградная, фенилмолочная
(миндальная), фенилуксусная, гиппуровая кислоты, фенилэтиламин,
фенилацетилглютамин) и вызванный ими ацидоз,
2) прямое токсическое действие указанных веществ на центральную
нервную
систему,
которое
заключается
в торможении фенилаланином активности ряда ферментов, в том
числе пируваткиназы (окисление глюкозы), тирозиназы (синтез
меланина), тирозингидроксилазы (синтез катехоламинов) и нарушение
синтеза моноаминовых нейромедиаторов – тирамина, октопамина,
3) нарушение синтеза серотонина, т.
 к. фенилаланин-4-монооксигеназа
к. фенилаланин-4-монооксигеназаодновременно осуществляет гидроксилирование триптофана до 5гидрокситриптофана,
предшественника
серотонина,
конкурентное снижение фенилаланином транспорта в клетки ароматических
аминокислот – триптофана и тирозина,
4) нарушение синтеза простых и сложных белков в тканях, что вызывает
тяжелые повреждения мозга и нарушение функции печени у большинства
больных.
Обмен АРГИНИНА
Аргинин — источник оксида азота (NO, ЭРФ) и
орнитина (АМК, не входящей в состав белков).
Состав NO-синтазы — гем, два флавиновых
кофермента (FAD и FMN), Н4БП, Zn2+ и Са2+
3 изоформы NO-синтаз — нейрональная,
эпителиальная и индуцибельная (миокард, печень, мышцы)
Оксид азота — сигнальная молекула, активирующая
гуанилатциклазу (стимуляция синтеза цГМФ).
Снижает силу сердечных сокращений, регулирует
тонус сосудов, тормозит апоптоз, предотвращает
агрегацию тромбоцитов, обладает
антиканцерогенной активностью.
NO:
— выступает как вторичный мессенджер и активирует
цитозольную гуанилатциклазу,
— является нейромедиатором,
— играет роль в регуляции сосудистого тонуса и расслаблении
гладкой мускулатуры сосудов,
— предотвращает агрегацию тромбоцитов и адгезию
нейтрофилов к эндотелию,
— обладает цитотоксической и микробицидной активностью.
Катаболизм АМК с
разветвленной цепью
(Вал, Лей, Илей) — не в печени, как у других АМК, а в
мышцах, жировой ткани, почках и в головном мозге.
Катаболизм проходит в 2 этапа:
1. Трансаминирование с -кетоглутаратом под
действием аминотрансферазы аминокислот с
разветвленной цепью — образуются -кетокислоты
2. Оксилительное декарбоксилирование кетокислот дегидрогеназным комплексом кетокислот с разветвленной цепью с образованием
ацил-КоА-производных
19. Катаболизм АМК с разветвленной цепью
Нарушение работыдегидрогеназного
комплексе -кислот
лежит в основе
заболеваний,
связанных с
нарушением
метаболизма
разветвленных
аминокислот –
болезнь
«кленового
сиропа».
Обмен триптофана — незаменимая АМК.
В физиологических условиях более 95%
триптофана окисляется по кинурениновому
пути и не более 1% — по серотониновому.
Триптофан
под
действием
гемсодержащего
фермента
триптофан-2,3-диоксигеназы
в
присутствии
молекулярного
кислорода
превращается в формил-кинуренин.

Формил-кинуренин является предшественником
рибонуклеотида никотиновой кислоты, участвует в
синтезе НАД, уменьшая потребность организма в
витамине РР.
Обмен дикарбоновых АМК
(глутаминовой и аспарагиновой)
Аспарагиновая кислота — участвует в
орнитиновом цикле мочевинообразования, в
реакции трансаминирования и биосинтезе
углеводов, карнозина и ансерина, пуриновых и
пиримидиновых нуклеотидов.
Глутаминовая кислота — служит (помимо
глюкозы) энергетическим материалом для
мозга, участвует в синтезе глутамина и
глутатиона.
Глутамин и Аспарагин подвергаются сочетанному
трансаминированию и
дезамидированию под
действием специфических
трансаминаз амидов
(глутаминтрансамидазы и
аспарагинтрансамидазы) и
неспецифической -амидазы.
English Русский Правила
Обмен аминокислот мышцами и печенью при сепсисе: сравнительные исследования in vivo и in vitro | JAMA Surgery
Обмен аминокислот мышцами и печенью при сепсисе: сравнительные исследования in vivo и in vitro | ДЖАМА Хирургия | Сеть ДЖАМА [Перейти к навигации]Эта проблема
- Скачать PDF
- Полный текст
- Поделиться
Твиттер Фейсбук Электронная почта LinkedIn
- Процитировать это
- Разрешения
Артикул
Февраль 1983 г.
Шимон Розенблатт, MD ; Джордж Х.А. Клоуз-младший, доктор медицины ; Барбара С. Джордж, MD ; и другие Эрвин Хирш, MD ; Бенгт Линдберг, MD
Принадлежность авторовОт отделений хирургии Гарвардской медицинской школы (Бостон) (доктор Розенблатт и Клоуз) и Медицинской школы Бостонского университета в больнице диакониссы Новой Англии (доктор Розенблатт, Клоуз, Джордж и Линдберг) и Бостонской городской больницы (доктор Розенблатт, Клоуз и Хирш).
Арка Сур. 1983;118(2):167-175. doi:10.1001/archsurg.1983.013
023004
Полный текст
Абстрактный
• «Центральная фракционная скорость клиренса» аминокислот (CFCR), отношение скорости поступления аминокислот во внеклеточный пул к размеру пула, является мерой поглощения и клиренса аминокислот печенью и другими висцеральными тканями. У девяти здоровых пациентов после абсорбции средний CFCR составил 5% по сравнению с 21% у 31 тяжело инфицированного пациента. В сравнительных целях были получены биоптаты печени и мышц для инкубации. У инфицированных пациентов скорость включения тирозина в белок в печени была в три раза выше, чем у неинфицированных пациентов, и хорошо коррелировала с CFCR. Не было существенной разницы в окислении тирозина в печени. В мышцах инфицированных пациентов общая деградация белка была в шесть раз выше, чем у неинфицированных пациентов. Инкубированные ткани крыс вели себя аналогичным образом. Таким образом, у людей с сепсисом, как и у животных, происходит ускоренный перенос аминокислот из мышц во внутренние органы для синтеза белка. CFCR продемонстрировал важность для выживания висцерального поглощения аминокислот; у выживших больных она составила 35%, и только у 19% среди умерших.
В сравнительных целях были получены биоптаты печени и мышц для инкубации. У инфицированных пациентов скорость включения тирозина в белок в печени была в три раза выше, чем у неинфицированных пациентов, и хорошо коррелировала с CFCR. Не было существенной разницы в окислении тирозина в печени. В мышцах инфицированных пациентов общая деградация белка была в шесть раз выше, чем у неинфицированных пациентов. Инкубированные ткани крыс вели себя аналогичным образом. Таким образом, у людей с сепсисом, как и у животных, происходит ускоренный перенос аминокислот из мышц во внутренние органы для синтеза белка. CFCR продемонстрировал важность для выживания висцерального поглощения аминокислот; у выживших больных она составила 35%, и только у 19% среди умерших.
( Arch Surg 1983;118:167-175)
Полный текст
Добавить или изменить учреждение
- Академическая медицина
- Кислотно-основное, электролиты, жидкости
- Аллергия и клиническая иммунология
- Анестезиология
- Антикоагулянты
- Искусство и образы в психиатрии
- Вспомогательная репродукция
- Кровотечение и переливание
- Кардиология
- Уход за тяжелобольным пациентом
- Проблемы клинической электрокардиографии
- Климат и здоровье
- Клиническая задача
- Поддержка принятия клинических решений
- Клинические последствия базовой нейронауки
- Клиническая фармация и фармакология
- Дополнительная и альтернативная медицина
- Заявления о консенсусе
- Коронавирус (COVID-19)
- Медицина интенсивной терапии
- Культурная компетентность
- Стоматология
- Дерматология
- Диабет и эндокринология
- Интерпретация диагностических тестов
- Разработка лекарств
- Электронные медицинские карты
- Скорая помощь
- Конец жизни
- Гигиена окружающей среды
- Справедливость, разнообразие и инклюзивность
- Этика
- Пластическая хирургия лица
- Гастроэнтерология и гепатология
- Генетика и геномика
- Геномика и точное здоровье
- Гериатрия
- Глобальное здравоохранение
- Руководство по статистике и методам
- Рекомендации
- Заболевания волос
- Модели медицинского обслуживания
- Экономика здравоохранения, страхование, оплата
- Качество медицинской помощи
- Реформа здравоохранения
- Медицинская безопасность
- Медицинские работники
- Различия в состоянии здоровья
- Несправедливость в отношении здоровья
- Информатика здравоохранения
- Политика здравоохранения
- Гематология
- История медицины
- Гуманитарные науки
- Гипертония
- Изображения в неврологии
- Наука внедрения
- Инфекционные болезни
- Инновации в оказании медицинской помощи
- Инфографика JAMA
- Право и медицина
- Ведущее изменение
- Меньше значит больше
- ЛГБТКИА Медицина
- Образ жизни
- Медицинский код
- Медицинские приборы и оборудование
- Медицинское образование
- Медицинское образование и обучение
- Медицинские журналы и публикации
- Меланома
- Мобильное здравоохранение и телемедицина
- Нарративная медицина
- Нефрология
- Неврология
- Неврология и психиатрия
- Примечательные примечания
- Сестринское дело
- Питание
- Питание, Ожирение, Упражнения
- Ожирение
- Акушерство и гинекология
- Гигиена труда
- Онкология
- Офтальмология
- Ортопедия
- Отоларингология
- Патология и лабораторная медицина
- Уход за пациентами
- Информация для пациентов
- Педиатрия
- Повышение производительности
- Показатели эффективности
- Периоперационный уход и консультации
- Фармакоэкономика
- Фармакоэпидемиология
- Фармакогенетика
- Фармация и клиническая фармакология
- Физическая медицина и реабилитация
- Физиотерапия
- Руководство врача
- Поэзия
- Здоровье населения
- Профессиональное благополучие
- Профессионализм
- Психиатрия и поведенческое здоровье
- Общественное здравоохранение
- Легочная медицина
- Радиология
- Регулирующие органы
- Исследования, методы, статистика
- Реанимация
- Ревматология
- Управление рисками
- Научное открытие и будущее медицины
- Совместное принятие решений и общение
- Медицина сна
- Спортивная медицина
- Трансплантация стволовых клеток
- Наркомания и наркология
- Хирургия
- Хирургические инновации
- Хирургический жемчуг
- Обучаемый момент
- Технологии и финансы
- Искусство JAMA
- Искусство и медицина
- Рациональное клиническое обследование
- Табак и электронные сигареты
- Токсикология
- Трансляционная медицина
- Травмы и травмы
- Приверженность лечению
- УЗИ
- Урология
- Руководство пользователя по медицинской литературе
- Вакцинация
- Венозная тромбоэмболия
- Здоровье ветеранов
- Насилие
- Женское здоровье
- Рабочий процесс и процесс
- Уход за ранами, инфекция, лечение
Сохранить настройки
Политика конфиденциальности | Условия использования
12.
 Метаболизм аминокислот • Функции клеток и организма человека
Метаболизм аминокислот • Функции клеток и организма человекаСодержание:
1. Введение в метаболизм белков и аминокислот
2. Деградация аминокислот
3. Деградация углеродного скелета аминокислот
4. Образование заменимых аминокислот в организме человека
5. Важные производные отдельных аминокислот
_
Введение в метаболизм белков и аминокислот
Белки являются наиболее важными и наиболее распространенными биомолекулами в организме человека – общий белок сумма соответствует 14 кг (верно для человека весом 70 кг). Совокупность всех аминокислот в организме называется пулом аминокислот . Как формируется этот пул? И как мы его используем?
Взрослый человек расщепляет примерно 300-500 г белков до аминокислот в день, это событие называется протеолизом . Примерно такое же количество аминокислот включается в белки в процессе, называемом протеосинтезом . Еще один источник аминокислот, белков, содержащихся в пище , что составляет примерно 70-100 г в день. Конечным источником аминокислот является пул биосинтеза из заменимых аминокислот образующие аминокислоты 30-40 г в день. В день расщепляется около 120 г аминокислот. В этом процессе аминокислотный скелет можно разделить на аминогруппы (-NH 2 и другие атомы азота) и углеродных цепи — каждая из них имеет совершенно другую судьбу, которая будет описана далее в этом разделе.
Конечным источником аминокислот является пул биосинтеза из заменимых аминокислот образующие аминокислоты 30-40 г в день. В день расщепляется около 120 г аминокислот. В этом процессе аминокислотный скелет можно разделить на аминогруппы (-NH 2 и другие атомы азота) и углеродных цепи — каждая из них имеет совершенно другую судьбу, которая будет описана далее в этом разделе.
Аминокислоты могут также служить предшественниками многих важных для организма веществ – напр. биогенные амины, гем или пурины и пиримидины.
Период полураспада белков заметно отличается от белка к белку и поэтому невозможно дать его среднее значение. Как правило, структурные белки более постоянны (и поэтому имеют более длительный период полураспада), в то время как молекулы многих ферментов существуют очень недолго – всего несколько десятков минут или часов.
Протеолиз — это полное расщепление белка до свободных аминокислот.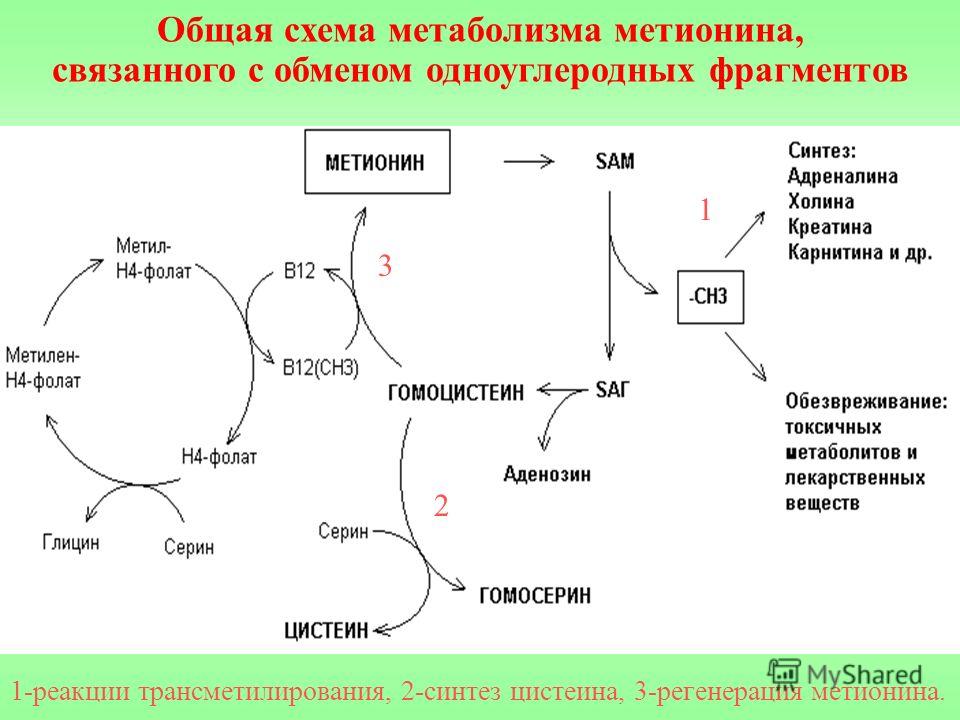 Протеазы и пептидазы (протеолитические ферменты) встречаются не только в желудочно-кишечного тракта , но и в каждой клетке – лизосом . Протеолитические ферменты классифицируются на:
Протеазы и пептидазы (протеолитические ферменты) встречаются не только в желудочно-кишечного тракта , но и в каждой клетке – лизосом . Протеолитические ферменты классифицируются на:
1) экзопептидазы – амино- и карбоксипептидазы – – ферменты, отщепляющие белки/пептиды от концов цепи
2) эндопептидазы – трипсин , химотрипсин или пепсин – являются ферментами, расщепляющими пептидные связи внутри белковых/пептидных цепей
Некоторые белки расщепляются на убиквитин-протеасомный комплекс . Убиквитин, небольшой белок, встречается во всех эукариотических клетках. Белок, предназначенный для деградации в протеасомах , помечен убиквитином. Этот процесс называется убиквитинированием (опционально полиубиквитинированием – если присутствует несколько молекул убиквитина).
Историческая корреляция:
Аарон Чехановер, Аврам Хершко и Ирвин Роуз получили Нобелевскую премию по химии (Открытие убиквитин-опосредованной деградации белка) в 2004 году.
Заменимые и незаменимые аминокислоты
Организм человека способен синтезировать только определенные аминокислоты (заменимые, незаменимые). Остальные аминокислоты (эссенциальные, незаменимые) должны поступать с пищей. Вот обзор:
Незаменимые аминокислоты
1) Разветвленные: Val, Leu, Ile
2) Ароматические: Phe, Trp
3) Основные: Lys 90 365
4) Серосодержащие: Met
5) С гидроксильной группой: Thr
Условно незаменимые аминокислоты
Arg, His
Заменимые аминокислоты
Gly, Ala, Ser, Pro, Cys, Tyr, Asn, Gln, Asp, Glu 9 0003
Важные реакции метаболизма аминокислот
1) Декарбоксилирование означает удаление карбоксильной группы – образуются биогенные амины
2) Трансаминирование означает обмен аминогруппы группа с 2-оксокислотой – Образуются 2-оксокислоты
3) Окислительное дезаминирование означает окислительное удаление аминогруппы – Образуются 2-оксокислоты
4) Пептидная связь образование – пептид и белок поколение
Дефекты метаболизма аминокислот
У человека существует множество различных генетических дефектов метаболизма аминокислот. Некоторые их промежуточные соединения накапливают в организме, вызывая дефект развития нервной системы что часто приводит к умственной отсталости .
Некоторые их промежуточные соединения накапливают в организме, вызывая дефект развития нервной системы что часто приводит к умственной отсталости .
_
Деградация аминокислот
Существует 20 (21, если включить селеноцистеин) основных протеиногенных аминокислот , которые могут быть вставлены в белковые молекулы в процессе трансляции. Катаболизм их углеродных скелетов покрывает примерно 10-15 % энергетических потребностей организма . Аминокислоты также могут служить субстратами (предшественниками) для биосинтеза других питательных веществ – углеводов (глюконеогенез) и липидов.
Теперь отдельно обсудим судьбу аминного азота и метаболизм аминокислотного углеродного скелета.
Удаление аминогруппы
Удаление аминогруппы является важным этапом катаболизма аминокислот. Азот аминогрупп (аминоазот) не может быть использован для производства энергии и должен быть удален из нашего организма. Первый способ – это превращение аминоазота в мочевину (около 95 % ) с последующим выведением мочевины из организма с мочой. Второй путь – высвобождение аминного азота из глютамина в форме 9.0364 NH 3 /NH 4 + в клетках канальцев почек (около 5 % ).
Первый способ – это превращение аминоазота в мочевину (около 95 % ) с последующим выведением мочевины из организма с мочой. Второй путь – высвобождение аминного азота из глютамина в форме 9.0364 NH 3 /NH 4 + в клетках канальцев почек (около 5 % ).
Трансаминирование
Трансаминирование представляет собой свободно обратимую реакцию, катализируемую трансаминазами ( аминотрансферазами ). Аминогруппа α-аминокислоты заменяется на оксогруппу 2-оксокислоты при трансаминировании – из аминокислоты получается 2-оксокислота, а из исходной 2-оксокислоты образуется аминокислота.
Аминогруппа переносится кофактором пиридоксальфосфатом ( PLP , производным витамина B6 ) на оксокислоту (образование основания Шиффа).
Большинство аминокислот подвергаются трансаминированию при их деградации . Ферменты аспартатаминотрансферазы (АСТ) и аланинаминотрансферазы (АЛТ) являются конкретными примерами трансаминаз, которые обычно выявляются как маркеры потенциального повреждения клеток печени. Катализируемые реакции АСТ и АЛТ представлены на схеме.
Катализируемые реакции АСТ и АЛТ представлены на схеме.
Образующиеся 2-оксокислоты (оксалоацетат и пируват) участвуют в энергетическом обмене внутри клеток.
Но есть исключения (например, треонин), который не расщепляется трансаминированием.
Превращение глутамата в глутамин
Превращение карбоксильной группы глутамата (в боковой цепи) в амидную группу глутамина катализируется цитозольным ферментом глутаминсинтетазой . АТФ и NH 4 + также необходимы. Эта реакция используется в клетках ЦНС как основной дезинтоксикационный механизм удаления токсичных NH 3 из тканей головного мозга. Возникающий глютамин является важнейшей транспортной формой аминного азота (аммиака) в крови – обеспечивает транспорт из внепеченочных тканей кровью в печень и почки. Глютамин имеет самую высокую концентрацию аминокислот в плазме – 0,6 ммоль/л (аланин – 0,3 ммоль/л). В молекуле глутамина «запасены» две аминогруппы/аммиака. Глутамин способен вводить аммиак в различные биосинтетические процессы – например, в синтез пуриновых оснований.
Глутамин способен вводить аммиак в различные биосинтетические процессы – например, в синтез пуриновых оснований.
Митохондриальный фермент глутаминаза катализирует высвобождение NH 3 из глутамина (гидролитическое дезаминирование, часто происходит в гепатоцитах и клетках почечных канальцев). Образующийся в митохондриях печени аммиак поступает в цикл мочевины. В почках аммиак выводится с мочой, где служит буфером.
Окислительное дезаминирование
При окислительном дезаминировании аминогруппа превращается в кетогруппу с одновременным выделением NH 3 . Глутамат – единственная аминокислота, которая дезаминируется с достаточной скоростью в организме человека. Глутаматдегидрогеназа , катализирующая окислительное дезаминирование глутамата , обнаружена в митохондриальном матриксе (в основном в клетках печени). Этот процесс демонстрирует следующую реакцию:
Глутамат + НАД + → α-кетоглутарат + NH 4 + + НАДН + Н +
Полученный NH 4 + вступает в цикл мочевины, а α-кетоглутарат может использоваться в трансаминировании или цикле Кребса. Указанная реакция полностью обратима – глутамат может быть синтезирован из α-KG и NH 4 + .
Указанная реакция полностью обратима – глутамат может быть синтезирован из α-KG и NH 4 + .
Можно сделать вывод, что большая часть аминокислоты подвергается трансаминированию при ее деградации и что большая часть амино-азота из аминокислот прямо или косвенно сосредоточена в молекуле глутамата/глутамина. Аминоазот впоследствии высвобождается в реакциях глутаминазы и глутаматдегидрогеназы.
Цикл мочевины (орнитина)
Токсичность аммиака
Аммиак – полярное вещество , свободно проходящее через физические барьеры , а также гематоэнцефалический барьер . При повышении его концентрации в организме нарушается баланс многих важных реакций. Рассмотрим следующие примеры:
Глутамат + НАД + → α-кетоглутарат + NH 4 +
Глутамат + NH 4 9 0383 + + АТФ → глутамин + АДФ + Pi
При избытке аммиака концентрация глутамина постепенно увеличивается . Но образование глютамина также потребляет α-кетоглутарат цикла Кребса – скорость этого пути постепенно снижается и соответственно производство энергии в клетках. Концентрация аммиака в плазме не должна превышать 35 мкмоль/л . В организме человека большая часть токсичного аммиака превращается в мочевину в результате реакций цикла мочевины.
Но образование глютамина также потребляет α-кетоглутарат цикла Кребса – скорость этого пути постепенно снижается и соответственно производство энергии в клетках. Концентрация аммиака в плазме не должна превышать 35 мкмоль/л . В организме человека большая часть токсичного аммиака превращается в мочевину в результате реакций цикла мочевины.
Реакции в цикле мочевины
Мочевина, нетоксичное соединение, транспортируется кровотоком в почки, где выводится с мочой. Цикл мочевины расположен в матриксе митохондрий и цитозоле клеток печени. Этот путь представляет собой энергозатратный процесс , в который вступают три субстрата – аммиак , углекислый газ (бикарбонат) и аспартат (его аминогруппа). Митохондриальная карбамоилфосфатсинтетаза I является регуляторным ферментом. Орнитиновый цикл связан с циклом Кребса через оксалоацетат и фумарат.
Образование мочевины включает пять реакций:
1) Образование карбамоилфосфата катализируется митохондриальной карбамоилфосфатсинтетазой I:
NH 4 905 55 + + HCO 3 – + АТФ → карбамоилфосфат + 2 АДФ + Pi
2) Образование цитруллина катализируется орнитинтранскарбамоилазой:
Орнитин + карбамоилфосфат → цитруллин + Pi
Цитруллин проходит в цитозоль.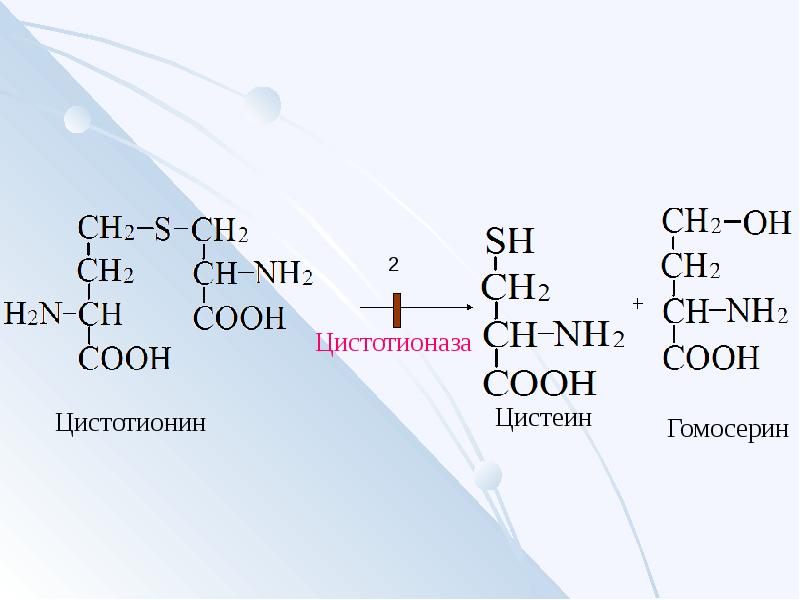
3) Образование аргининосукцината катализируется аргининосукцинатсинтетазой:
Цитруллин + Asp + АТФ → аргининосукцинат + AMP + PPi
4) Расщепление аргининосукцината 90 365 катализируется аргининосукцинатлиазой:
Аргининосукцинат → аргинин + фумарат
5) Гидролиз аргинина катализируется аргиназой:
Аргинин + H 2 O → орнитин + мочевина
Орнитин возвращается в митохондриальный матрикс.
Цикл мочевины тесно связан с циклом Кребса – из возникающего фумарата становится аспартат. Как работают эти отношения? Фумарат сначала гидратируется до малата, который путем окисления превращается в оксалоацетат. Фермент аспартатаминотрансферазы катализирует трансаминирование между глутаматом и оксалоацетатом, в результате чего аспартат входит в орнитиновый цикл. Глутамат получают трансаминированием расщепленных аминокислот, которые передают свои аминогруппы на молекулу α-кетоглутарата.
Регуляция орнитинового цикла
Карбамоилфосфатсинтетаза I , основной регуляторный фермент орнитинового цикла, активируется N-ацетилглутаматом. Фермент N-ацетилглутаматсинтетаза катализирует реакцию между AcCoA и глутаматом, в результате которой образуется N-ацетилглутамат. Аминокислота аргинин повышает активность фермента. Транскрипция ферментов цикла мочевины увеличивается при высокобелковой диете или увеличивает катаболизм белка (например, голодание), поэтому в повышенном поступлении аминокислот. Цикл мочевины относится к протонпродуцирующим реакциям, его активность снижается при более низких значениях рН – ацидоз.
Глюкозо-аланиновый цикл
Аланин участвует в передаче аммиака кровью , а также служит через пируват важным источником углерода в глюконеогенезе – см. подраздел 2/9. Глюкозо-аланиновый цикл представляет собой путь, проходящий между печенью и мышечные клетки .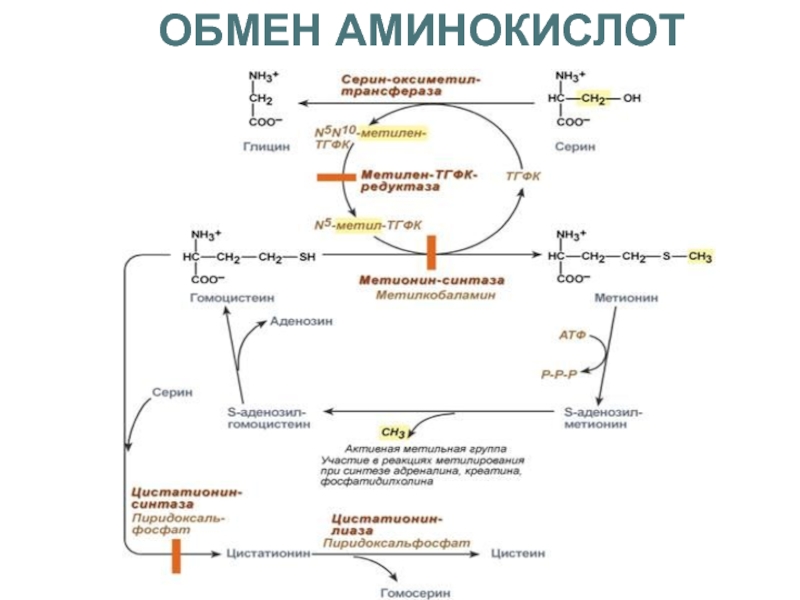 Пируват, образующийся в мышечных клетках, подвергается переаминированию с образованием аланина. Он высвобождается в кровь и переносится в печень, где снова превращается в пируват путем переаминирования, которое может участвовать в процессе глюконеогенеза. Образовавшаяся глюкоза высвобождается в кровь и поступает в мышечные клетки, и цикл продолжается. Передаваемая аминогруппа (аммиак) направляется в цикл мочевины.
Пируват, образующийся в мышечных клетках, подвергается переаминированию с образованием аланина. Он высвобождается в кровь и переносится в печень, где снова превращается в пируват путем переаминирования, которое может участвовать в процессе глюконеогенеза. Образовавшаяся глюкоза высвобождается в кровь и поступает в мышечные клетки, и цикл продолжается. Передаваемая аминогруппа (аммиак) направляется в цикл мочевины.
_
Деградация углеродных скелетов аминокислот
Белки в организме человека содержат 20 (21, если включить селеноцистеин) протеиногенных аминокислот. Для катаболизма углеродных скелетов аминокислот существует двадцать (двадцать одна) различных мультиферментных последовательностей. В этом тексте мы ограничимся лишь основными общими механизмами деградации углеродного скелета аминокислот и несколькими избранными примерами.
Катаболизм углеродных скелетов аминокислот приводит к образованию семи продуктов: пируват , ацетил-КоА , ацетоацетил-КоА, α-кетоглутарат , сук-КоА , фумарат и оксалоацетат .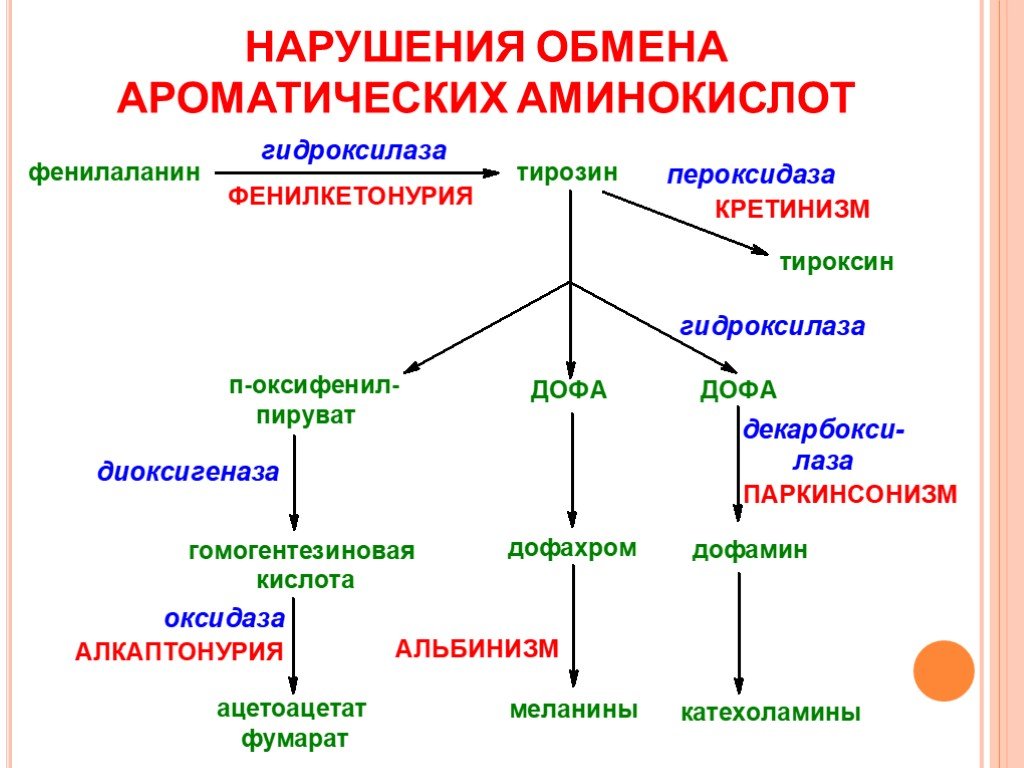 У них разная судьба в энергетическом обмене. Стратегия клетки заключается в преобразовании углеродных скелетов аминокислот в соединения, полезные для глюконеогенеза, или в молекулы липидов (жирные кислоты и кетоновые тела). Аминокислоты делятся на глюкогенные и кетогенные аминокислоты в зависимости от судьбы продуктов их распада. Аминокислоты лейцин и лизин (начинающиеся с буквы L ) относятся к кетогенным аминокислотам, которые приводят к образованию ацетил-КоА и ацетоацетил-КоА. К глюкогенным аминокислотам относятся те, которые приводят к образованию остальных пяти продуктов – пируват , α-кетоглутарат , сук-КоА , фумарат или оксалоацетат – серин, треонин, цистеин, метионин, аспартат, глутамат, аспарагин, глутамин, глицин, аланин, валин, пролин, гистидин и аргинин. Но некоторые аминокислоты имеют два продукта деградации — один из них глюкогенный, а второй — кетогенный. Эти аминокислоты называются 9.
У них разная судьба в энергетическом обмене. Стратегия клетки заключается в преобразовании углеродных скелетов аминокислот в соединения, полезные для глюконеогенеза, или в молекулы липидов (жирные кислоты и кетоновые тела). Аминокислоты делятся на глюкогенные и кетогенные аминокислоты в зависимости от судьбы продуктов их распада. Аминокислоты лейцин и лизин (начинающиеся с буквы L ) относятся к кетогенным аминокислотам, которые приводят к образованию ацетил-КоА и ацетоацетил-КоА. К глюкогенным аминокислотам относятся те, которые приводят к образованию остальных пяти продуктов – пируват , α-кетоглутарат , сук-КоА , фумарат или оксалоацетат – серин, треонин, цистеин, метионин, аспартат, глутамат, аспарагин, глутамин, глицин, аланин, валин, пролин, гистидин и аргинин. Но некоторые аминокислоты имеют два продукта деградации — один из них глюкогенный, а второй — кетогенный. Эти аминокислоты называются 9. 0364 кето- и глюкогенные аминокислоты – к ним относятся изолейцин, фенилаланин, тирозин и триптофан.
0364 кето- и глюкогенные аминокислоты – к ним относятся изолейцин, фенилаланин, тирозин и триптофан.
В следующем обзоре показаны продукты деградации определенных аминокислот:
1) Ацетил-КоА и ацетоацетил-КоА – Lys и Leu являются чисто кетогенными аминокислотами, некоторые другие аминокислоты (Phe, Tyr, Trp, Ile) дают глюкогенные и кетогенные продукты деградации
90 364 2) α-кетоглутарат – пятиуглеродные аминокислоты – Glu, Gln, Pro, Arg a His
3) Suc-CoA – неполярные аминокислоты – Met, Ile a Val
4) Фумарат – Phe, Tyr
5) Оксалоацетат – четырехуглеродные аминокислоты – As p a Asn
6) Пируват – Cys, Ala, Ser, Gly, Thr, Trp
Деградация разветвленных аминокислот – Val, Leu и Ile
Эти аминокислоты не деградируют в клетках печени, а преимущественно во внепеченочных тканях – особенно высокая активность у мышечные клетки . Они содержат определенные трансаминаза , продуцирующая соответствующие α-кетокислоты – так называемые кетоаналоги разветвленных аминокислот . Эта трансаминаза отсутствует в клетках печени. Кетоаналоги превращаются в производные ацил-КоА с помощью комплекса дегидрирования , который катализирует окислительное декарбоксилирование и дегидрирование .
Эта трансаминаза отсутствует в клетках печени. Кетоаналоги превращаются в производные ацил-КоА с помощью комплекса дегидрирования , который катализирует окислительное декарбоксилирование и дегидрирование .
Генетический дефект комплекса дегидрирования вызывает болезнь кленового сиропа мочи . Это относительно редкое заболевание может приводить к накоплению соответствующих α-кетокислот в тканях и жидкостях организма (моча пахнет карамелью). Дефект вызывает аномальное развитие мозга, умственную отсталость и может привести к смерти человека.
_
Образование заменимых аминокислот в организме человека
Организм человека не способен синтезировать незаменимые аминокислоты – Phe, Trp, Val, Leu, Ile, Met, Thr и Lys. Две аминокислоты незаменимы в росте организма (период их повышенного потребления), скорость их синтеза недостаточна для покрытия потребностей организма – так называемые условно незаменимые аминокислоты – Arg, His.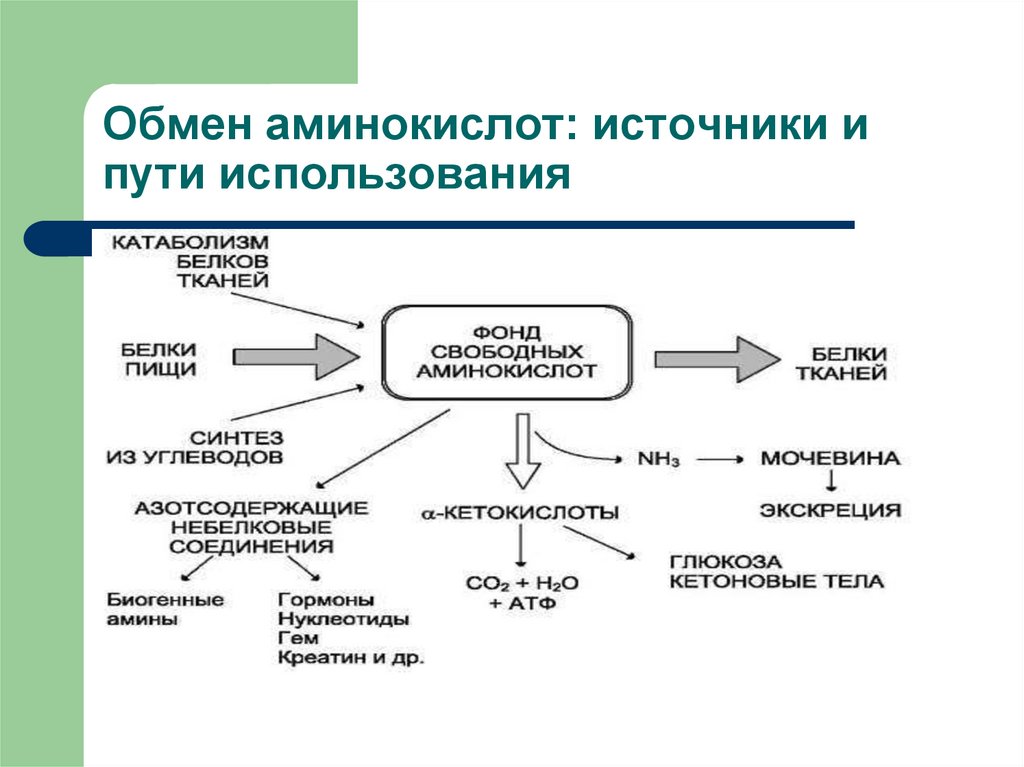 Остальные аминокислоты относятся к заменимым аминокислотам. Вот обзор предшественников аминокислот:
Остальные аминокислоты относятся к заменимым аминокислотам. Вот обзор предшественников аминокислот:
1) Оксалоацетат → Asp , Asn
2) α-кетоглутарат → Glu , Gln , Pro , ( Arg и His )
3) Пируват → Ala
4) 3-фосфоглицерат → Ser , Cy s и Gly
5) Phe → Tyr
Фенилкетонурия (ФКУ)
Фенилкетонурия (ФКУ) является аутосомно-рецессивным нарушением обмена веществ (заболеваемость 8-10 случаев/100 000 человек) обусловлено отсутствием или снижением активности фенилаланингидроксилазы. Физиологически этот фермент катализирует гидроксилирование Phe до Tyr. Дефектный фермент приводит к накоплению фенилаланина и альтернативной деградации Phe – образуются фенилпируват (трансаминирование), фениллактат, фенилацетат и фенилэтиламин . Такие вещества накапливаются в тканях и жидкостях организма и вырабатывают типичный запах мочи (мышиный запах) . Некоторые из них вызывают тяжелых повреждения головного мозга . Фенилкетонурия была первым обнаруженным генетическим дефектом метаболизма аминокислот у человека и в настоящее время является одним из заболеваний, для которых проводится скрининг всех новорожденных . Если он обнаружен даже в этом возрасте, мы можем предотвратить повреждение головного мозга с помощью специальной диеты с низким содержанием Phe .
Некоторые из них вызывают тяжелых повреждения головного мозга . Фенилкетонурия была первым обнаруженным генетическим дефектом метаболизма аминокислот у человека и в настоящее время является одним из заболеваний, для которых проводится скрининг всех новорожденных . Если он обнаружен даже в этом возрасте, мы можем предотвратить повреждение головного мозга с помощью специальной диеты с низким содержанием Phe .
_
Важные производные отдельных аминокислот
Декарбоксилирование – образование биогенных аминов
Некоторые аминокислоты подвергаются декарбоксилированию ( удаление карбоксильной группы ). В результате образуются биогенные амины (моноамины), обладающие широким спектром функций в организме человека. Вот основная схема:
1) Tyr → катехоламины (DOPA → дофамин → норадреналин (норэпинефрин) → адреналин (эпинефрин)
2) Trp → серотонин (5-гидрокситриптамин)
90 002 3) Glu → γ-аминомасляная кислота ( ГАМК )4) Гис → гистамин
5) Сер → этаноламин → холин → ацетилхолин
6) Cys → цистеамин
7) Asp → β-аланин
Оксид азота (NO)
Оксид азота является сосудорасширяющим веществом , вырабатываемым эндотелиальными клетками.
