Гликоген: құрылымы, синтезі, деградациясы, функциялары — Ғылым
Вызшақ: ГЕНДІК МУТАЦИЯЛАР. ГЕНДІК АУРУЛАР. БИОХИМИЯЛЫҚ ӘДІСМазмұны
- Құрылым
- Синтез
- Деградация
- Синтез бен деградацияны реттеу
- Синтез туралы
- Деградация
- Мүмкіндіктер
- Осыған байланысты аурулар
- Таңдаулы мысалдар
- Әдебиеттер тізімі
The гликоген бұл көптеген сүтқоректілердің көмірсуы. Көмірсулар әдетте қанттар деп аталады және оларды гидролизден туындаған қалдықтар санына қарай жіктейді (моносахаридтер, дисахаридтер, олигосахаридтер және полисахаридтер).
Моносахаридтер — олардың құрамына кіретін көміртектер санына қарай жіктелетін қарапайым көмірсулар. Одан кейін триосалар (3C), тетрозалар (4C), пентозалар (5C), гексозалар (6C), гептозалар (7C) және октозалар (8C) бар.
Альдегид тобының немесе кетон тобының болуына байланысты бұл моносахаридтер сәйкесінше альдозалар немесе кетозалар болып жіктеледі.
Дисахаридтер гидролиз арқылы екі қарапайым моносахаридті тудырады, ал олигосахаридтер 2 ден 10 моносахарид бірлігін, ал полисахаридтер 10 данадан көп моносахарид түзеді.
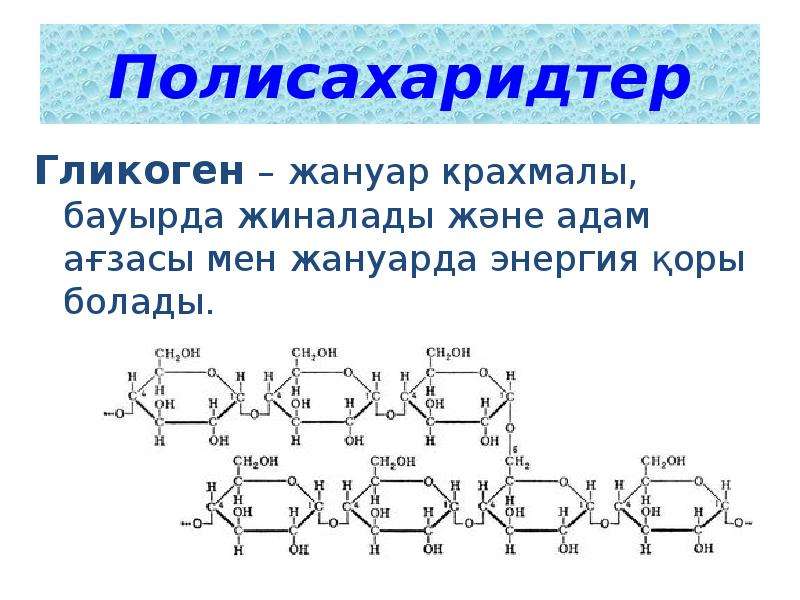
Гликоген — биохимиялық тұрғыдан алғанда алты көміртекті альдозаның тармақталған тізбектерінен тұратын полисахарид, яғни глюкоза деп аталатын гексоза. Гликогенді глюкоза ағашы ретінде графикалық түрде ұсынуға болады. Мұны жануарлар крахмалы деп те атайды.
Өсімдіктердегі глюкоза крахмал түрінде, ал жануарларда гликоген ретінде, ең алдымен бауыр мен бұлшықет тінінде сақталады.
Бауырда гликоген массасының 10% және бұлшықет массасының 1% құрауы мүмкін. 70 кг салмақтағы бауырдың салмағы шамамен 1800 г, бұлшық еттері 35 кг сияқты, бұлшықет гликогенінің жалпы мөлшері бауырға қарағанда әлдеқайда көп.
ҚұрылымГликогеннің молекулалық салмағы 108 г / мольға жетуі мүмкін, бұл 6 × 105 глюкоза молекулаларына тең. Гликоген α-D-глюкозаның бірнеше тармақталған тізбектерінен тұрады. Глюкоза (C6h22O6) — сызықтық немесе циклдік түрде ұсынылатын альдогексоза.
Гликоген α-D-глюкозаның бірнеше тармақталған тізбектерінен тұрады. Глюкоза (C6h22O6) — сызықтық немесе циклдік түрде ұсынылатын альдогексоза.
Гликоген α- (1 → 4) глюкозидтік байланыстармен байланысқан α-D-глюкоза түріндегі глюкозаның 12-ден 14-ке дейінгі қалдықтары бар тізбектері бар өте тармақталған және ықшам құрылымға ие. Тізбектің тармақтары α- (1 → 6) глюкозидтік байланыстар арқылы түзіледі.
Гликоген, диетадағы крахмал сияқты, ағзаға қажет көмірсулардың көп бөлігін қамтамасыз етеді. Ішекте бұл полисахаридтер гидролиз арқылы ыдырайды, содан кейін қанға негізінен глюкоза ретінде сіңеді.
Үш фермент: ß-амилаза, α-амилаза және амил-α- (1 → 6) -глюкозидаза гликогеннің де, крахмалдың да ішектің ыдырауына жауап береді.
Α-Амилаза кездейсоқ түрде гликогеннің және крахмалдың бүйір тізбектерінің α- (1 → 4) байланыстарын гидролиздейді, сондықтан эндогликозидаза деп аталады. Ss-амилаза — экзогликозидаза, бұтақтарға жетпей, шеткі тізбектердің ұштарынан α- (1 → 4) гликозидтік байланыстарды үзу арқылы ß-мальтоза димерлерін шығарады.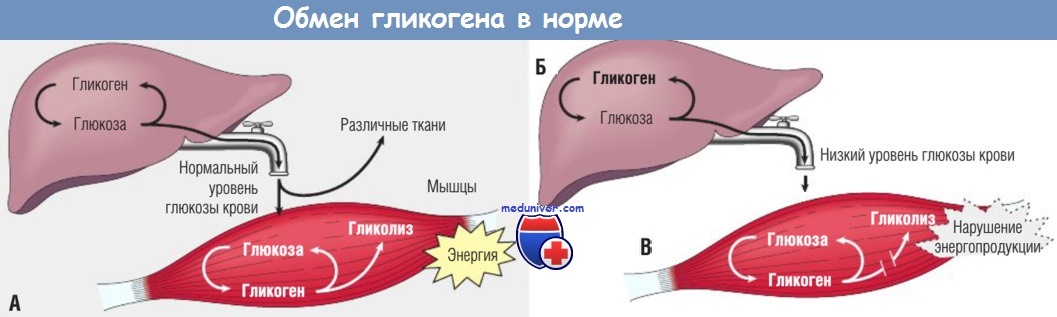
Ss-амилаза да, α-амилаза да тармақталған бөлшектерді бұзбайтын болғандықтан, олардың әсер етуінің соңғы өнімі шекаралас декстрин деп аталатын глюкозаның шамамен 35-тен 40-қа дейінгі қалдықтары бар жоғары тармақталған құрылым болып табылады.
Шектік декстрин ақырында α- (1 → 6) байланысы бар тармақтық нүктелерде амил-α- (1 → 6) -глюкозидаза көмегімен гидролизденеді, оны «азайтқыш» фермент деп те атайды. Одан кейін босатылған тізбектер ß-амилаза және α-амилаза әсерінен ыдырайды.
Жұтылған гликоген глюкоза ретінде енетін болғандықтан, ұлпаларда кездесетін зат организмге глюкозадан синтезделуі керек.
СинтезГликоген синтезі гликогенез деп аталады және негізінен бұлшықет пен бауырда жүреді. Денеге диетамен бірге кіретін глюкоза қанға, одан клеткаларға өтіп, глюкокиназа деп аталатын ферменттің әсерінен бірден фосфорланады.
Глюкокиназа глюкозаны көміртегі 6-да фосфорландырады. АТФ бұл реакция үшін фосфор мен энергияны қамтамасыз етеді. Нәтижесінде глюкоза 6-фосфат түзіліп, АДФ бөлінеді. Содан кейін глюкоза 6-фосфат фосфорды 6-позициядан 1-позицияға жылжытатын фосфоглукомутазаның әсерінен глюкозаның 1-фосфатына айналады.
Содан кейін глюкоза 6-фосфат фосфорды 6-позициядан 1-позицияға жылжытатын фосфоглукомутазаның әсерінен глюкозаның 1-фосфатына айналады.
Глюкоза 1-фосфат басқа үш ферменттер жиынтығының қатысуын көздейтін гликоген синтезі үшін белсендіріледі: UDP-глюкоза пирофосфорилаза, гликоген синтетаза және амил- (1,4 → 1,6) -гликозилтрансфераза.
Глюкоза-1-фосфат уридин трифосфатымен (UTP, уридин трифосфатының нуклеозиди) және UDP-глюкоза-пирофосфорилазаның әсерінен уридин дифосфат-глюкоза кешенін (UDP Glc) құрайды. Процесс барысында пирофосфат ионы гидролизденеді.
Содан кейін гликоген синтетаза ферменті UDP Glc комплексінің C1 және гликогеннің глюкозаның терминалдық қалдықтарының C4 арасында гликозидтік байланыс түзеді, ал UDP активтендірілген глюкоза кешенінен шығады. Бұл реакцияның болуы үшін «алғашқы гликоген» деп аталатын гликоген молекуласы болуы керек.
Алғашқы гликоген 37 кДа құрайтын және UDP Glc кешені арқылы тирозин қалдықтарына дейін гликозилденетін праймерлік протеин — гликогенинде синтезделеді. Ол жерден α-D-Глюкозаның қалдықтары 1 → 4 байланыстармен байланысады және гликоген синтетаза әсер ететін шағын тізбек түзіледі.
Ол жерден α-D-Глюкозаның қалдықтары 1 → 4 байланыстармен байланысады және гликоген синтетаза әсер ететін шағын тізбек түзіледі.
Бастапқы тізбек кем дегенде 11 глюкозаның қалдықтарын байланыстырғаннан кейін, тармақталатын фермент немесе амил- (1,4 → 1,6) -гликозилтрансфераза 6 немесе 7 глюкозаның қалдықтарынан тұратын тізбектің бөлігін 1 позициясында іргелес тізбекке береді. → 6, осылайша филиал пунктін құру. Осылайша құрылған гликоген молекуласы 1 → 4 гликозидтік байланысы бар глюкоза бірліктерін және одан да көп тармақтарды қосқанда өседі.
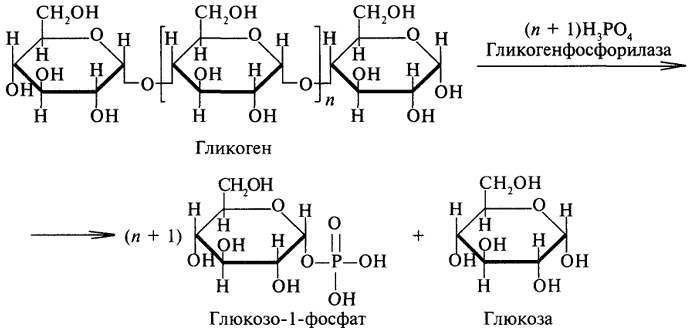
ДеградацияГликогеннің ыдырауы гликогенолиз деп аталады және бұл оның синтезінің кері жолына тең келмейді. Бұл жолдың жылдамдығы гликоген фосфорилаза катализденетін реакция жылдамдығымен шектеледі.
Гликоген фосфорилазы глюкоза 1-фосфатын бөліп, гликоген тізбегінің 1 → 4 байланысының бөлінуіне (фосфоролиз) жауап береді. Ферменттердің әрекеті шеткі тізбектердің ұштарынан басталады және бұтақтардың әр жағында 4 глюкозаның қалдықтары қалмайынша олар дәйекті түрде жойылады.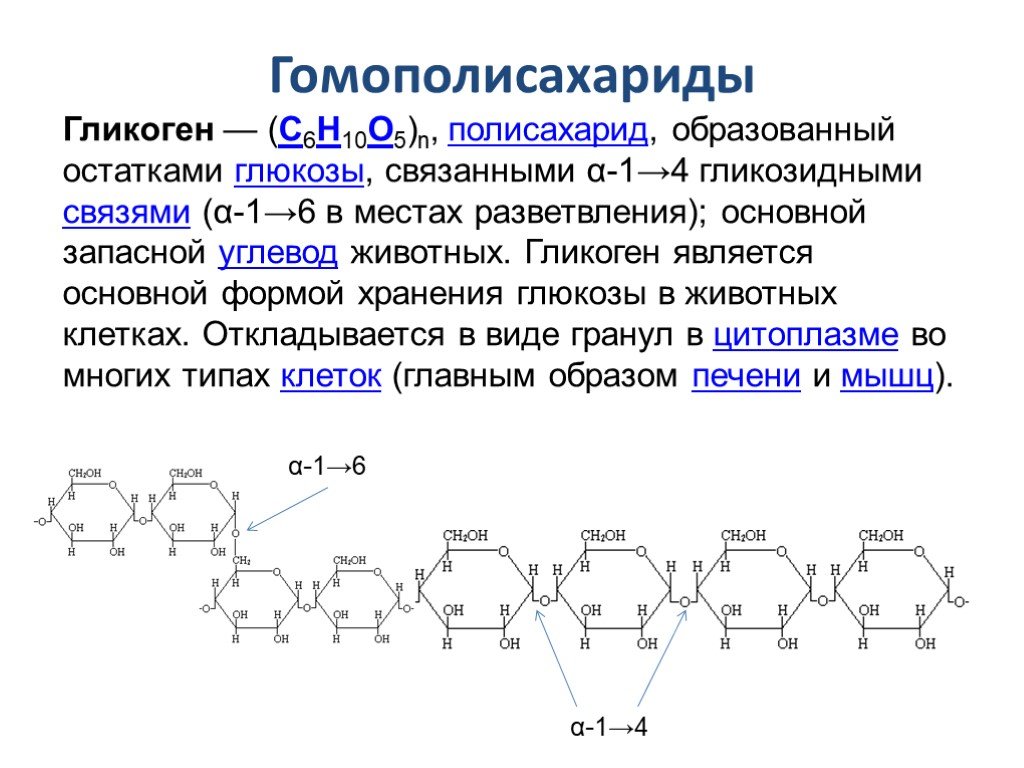
Содан кейін тағы бір фермент α- (1 → 4) → α- (1 → 4) глюкан трансферазы трисахарид бірлігін екінші бұтақтан екіншісіне ауыстыру арқылы тармақталған нүктені ашады. Бұл амил- (1 → 6) -глюкозидазаға (ферментті азайту) фосфорилаза әсерінен өтетін тармақты алып тастап, 1 → 6 байланысын гидролиздеуге мүмкіндік береді. Осы ферменттердің бірлескен әрекеті гликогенді толығымен бөлшектеуге аяқталады.
Бастапқы фосфомутаза реакциясы қайтымды болғандықтан, глюкозаның бөлінген глюкоза 1-фосфат қалдықтарынан глюкоза 6-фосфат түзілуі мүмкін. Бауыр мен бүйректе, бірақ бұлшық еттерінде емес, глюкоза-6-фосфатаза депосфосфорландырып, оны бос глюкозаға айналдыруға қабілетті фермент — глюкоза-6-фосфатаза бар.
Дефосфорланған глюкоза қанға диффузиялануы мүмкін, сондықтан бауыр гликогенолизі қандағы глюкоза (гликемия) деңгейінің жоғарылауында көрінеді.
Синтез бен деградацияны реттеуСинтез туралыБұл процесс екі негізгі ферменттерге әсер етеді: гликоген синтетаза және гликогенфосфорилаза, олардың біреуі белсенді болғанда, екіншісі оның белсенді емес күйінде болады.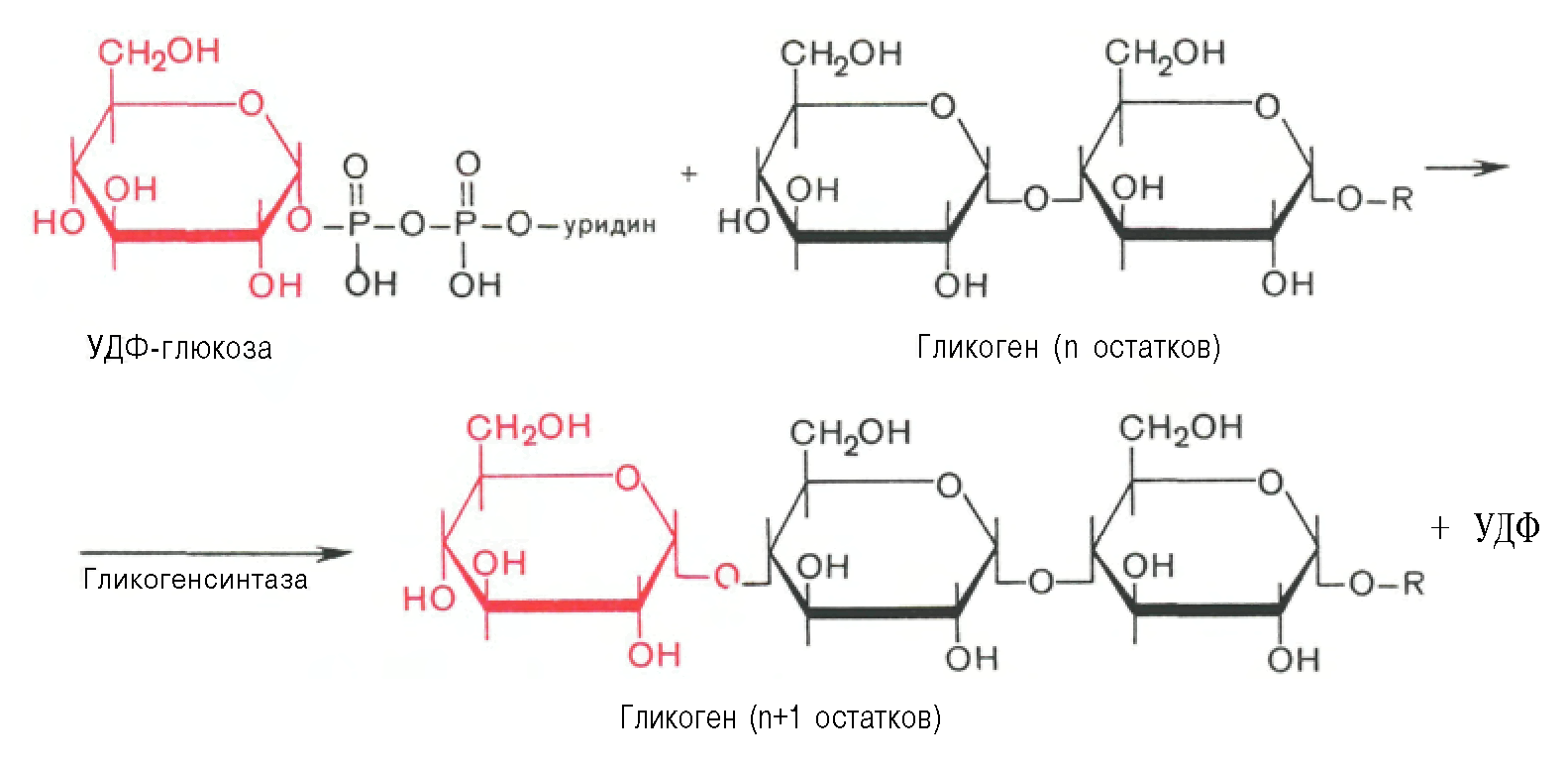 Бұл реттеу синтез бен ыдыраудың қарама-қарсы реакцияларының қатар жүруіне жол бермейді.
Бұл реттеу синтез бен ыдыраудың қарама-қарсы реакцияларының қатар жүруіне жол бермейді.
Екі ферменттердің белсенді формасы мен белсенді емес формасы бір-бірінен өте ерекшеленеді, ал фосфорилаза мен гликоген синтетазаның белсенді және енжар формаларының өзара конверсиясы қатаң гормоналды бақыланады.
Адреналин — бұл бүйрек үсті безінен шығатын гормон, ал глюкагон — ұйқы безінің эндокриндік бөлігінде түзілетін басқа зат. Эндокриндік ұйқы безі инсулин мен глюкагон шығарады. Лангерганс аралдарының α жасушалары глюкагонды синтездейтіндер.
Адреналин мен глюкагон — бұл қандағы глюкоза деңгейінің төмендеуіне жауап беру үшін энергия қажет болған кезде бөлінетін екі гормон. Бұл гормондар гликоген фосфорилазаның активтенуін ынталандырады және гликоген синтетазаны тежейді, осылайша гликогенолизді ынталандырады және гликогенезді тежейді.
Адреналин бұлшықет пен бауырға әсер етсе, глюкагон тек бауырға әсер етеді. Бұл гормондар аденилатциклазаны белсендіретін мақсатты жасушадағы арнайы мембраналық рецепторлармен байланысады.
Аденилатциклазаның активациясы ферментативті каскадты бастайды, ол бір жағынан гликоген синтетазаны инактивациялайтын және гликогенфосфорилазаны фосфорлану жолымен активтендіретін (сәйкесінше, жанама түрде) камп-тәуелді протеинкиназаны белсендіреді.
Қаңқа бұлшықетінде гликоген фосфорилазаның кальций арқылы активтенуінің тағы бір механизмі бар, ол жиырылу басында бұлшықет қабығының деполяризациясы нәтижесінде бөлінеді.
ДеградацияБұрын сипатталған ферменттік каскадтар глюкоза деңгейінің жоғарылауымен аяқталады және олар белгілі бір деңгейге жеткенде гликогенез белсендіріліп, гликогенолиз тежеледі, сонымен қатар эпинефрин мен глюкагонның кейінгі шығуы тежеледі.
Гликогенез гликоген синтезінің ингибиторы болып табылатын фосфорилаза киназа мен α фосфорилазаны инактивациялауды қамтитын әр түрлі механизмдермен гликоген синтезін реттейтін фермент — фосфорилаза фосфатазаның активациясы арқылы активтенеді.
Инсулин глюкозаның бұлшықет жасушаларына енуіне ықпал етеді, глюкозаның 6-фосфат деңгейін жоғарылатады, бұл гликоген синтетазаның депосфорилденуін және активтенуін ынталандырады. Осылайша синтез басталып, гликогеннің ыдырауы тежеледі.
Осылайша синтез басталып, гликогеннің ыдырауы тежеледі.
Бұлшықет гликогені бұлшықет үшін энергия қорын құрайды, ол резервтік майлар сияқты бұлшықетке өз функцияларын орындауға мүмкіндік береді. Глюкозаның көзі бола отырып, бұлшықет гликогені жаттығу кезінде қолданылады. Бұл резервтер дене шынықтырумен бірге артады.
Бауырда гликоген сонымен қатар ағзаның қызметі үшін де, дененің қалған бөлігін глюкозамен қамтамасыз ету үшін де маңызды қор болып табылады.
Бауыр гликогенінің бұл қызметі бауырда глюкоза 6-фосфаттан фосфат тобын алып тастауға және оны бос глюкозаға айналдыруға қабілетті фермент — глюкоза 6-фосфатаза болатындығымен байланысты. Бос глюкоза, фосфорланған глюкозадан айырмашылығы, гепатоциттердің (бауыр жасушаларының) мембранасы арқылы диффузиялануы мүмкін.
Осылайша бауыр ұзақ уақыт ашығу жағдайында да қан айналымын глюкозамен қамтамасыз етіп, глюкозаның тұрақты деңгейін сақтай алады.
Бұл функцияның маңызы өте зор, өйткені ми тек қана қандағы глюкозаға сүйенеді, сондықтан ауыр гипогликемия (қандағы глюкозаның өте төмен концентрациясы) сананың жоғалуына әкелуі мүмкін.
Гликогенмен байланысты ауруларды жалпылама түрде «гликогенді сақтау аурулары» деп атайды.
Бұл аурулар тіндерге қалыптан тыс мөлшерде немесе гликогеннің түсуімен сипатталатын тұқым қуалайтын патологиялар тобын құрайды.
Гликогенді сақтау ауруларының көпшілігі гликоген метаболизміне қатысатын кез-келген ферменттердегі генетикалық табиғат тапшылығынан туындайды.
Олар сегіз түрге жіктеледі, олардың көпшілігінің өз атаулары бар және олардың әрқайсысы әр түрлі фермент тапшылығынан туындайды. Кейбіреулері өмірдің өте ерте кезеңінде өледі, ал басқалары жаттығулар кезінде бұлшықет әлсіздігімен және тапшылығымен байланысты.
Таңдаулы мысалдарГликогенмен байланысты ең танымал аурулардың кейбіреулері:
— Фон Джирке ауруы немесе гликогенді сақтаудың І типті ауруы бауыр мен бүйректегі глюкозаның 6-фосфатаза тапшылығынан туындайды.
Бұл гликоген мен гипогликемияның шамадан тыс жинақталуына байланысты бауырдың қалыптан тыс өсуімен сипатталады (гепатомегалия), өйткені бауыр айналымға глюкозаны жеткізе алмайды. Мұндай аурумен ауыратын науқастардың өсуі бұзылады.
Мұндай аурумен ауыратын науқастардың өсуі бұзылады.
— Помпе немесе II типті ауру бауырда, жүрек пен қаңқа бұлшықеттерінде α- (1 → 4) -глюкан 6-гликозилтрансфералардың жетіспеуінен болады. Бұл ауру, Андерсен немесе IV типтегі сияқты, екі жасқа дейін өліммен аяқталады.
— McArdle немесе V типті ауру бұлшықет фосфорилазының жетіспеушілігін көрсетеді және бұлшықет әлсіздігімен, жаттығуға төзімділіктің төмендеуімен, бұлшықет гликогенінің қалыптан тыс жинақталуымен және жаттығу кезінде лактаттың жетіспеушілігімен бірге жүреді.
Әдебиеттер тізімі- Бхаттачария, К. (2015). Бауыр гликогенін сақтау ауруларын зерттеу және басқару. Аудармалы педиатрия, 4(3), 240–248.
- Dagli, A., Sentner, C., & Weinstein, D. (2016). III типтегі гликогенді сақтау ауруы. Ген-шолулар, 1–16.
- Guyton, A., & Hall, J. (2006). Медициналық физиология оқулығы (11-ші басылым). Elsevier Inc.
- Mathews, C., van Holde, K., & Ahern, K.
 (2000). Биохимия (3-ші басылым). Сан-Франциско, Калифорния: Пирсон.
(2000). Биохимия (3-ші басылым). Сан-Франциско, Калифорния: Пирсон. - Маккиернан, П. (2017). Бауыр гликогенін сақтау ауруларының патобиологиясы. Curr Pathobiol Rep.
- Мюррей, Р., Бендер, Д., Ботэм, К., Кеннелли, П., Родвелл, В., & Вайл, П. (2009). Харпердің иллюстрацияланған биохимиясы (28-ші басылым). McGraw-Hill медициналық.
- Nelson, D. L., & Cox, M. M. (2009). Лехингер Биохимияның принциптері. Омега шығарылымдары (5-ші басылым).
- Роун, Дж. Д. (1998). Биохимия. Берлингтон, Массачусетс: Нил Паттерсонның баспагерлері.
- Тарнопольский, М.А. (2018). Гликоген метаболизмінің бұзылуына байланысты миопатиялар. Нейротерапевтика.
Гликоген дегеніміз не? Ол неде жинақталған және оның физикалық күш салуға қандай қатысы бар? – Менің қасымда пайдалы тағам
Гликоген — полисахарид немесе полисахарид. Ол көптеген глюкоза молекулаларынан тұрады және жаттығу кезінде осы пішінге бөлінуі мүмкін. Күшті жаттығулар кезінде гликоген глюкозаның қарапайым түріне дейін ыдырайды. Ағза басқа заттармен қатар полисахаридтерге айналатын көмірсулармен қамтамасыз етілетінін есте ұстаған жөн, олар өз кезегінде бұлшықеттерде және ең бастысы — бауырда сақталады.
Ағза басқа заттармен қатар полисахаридтерге айналатын көмірсулармен қамтамасыз етілетінін есте ұстаған жөн, олар өз кезегінде бұлшықеттерде және ең бастысы — бауырда сақталады.
Гликоген және физикалық белсенділік
Спортзалда физикалық белсенді болуды елестетіп көріңіз. Сонда біздің бұлшық еттердің көп бөлігі қатысады, және сіз білетіндей, энергия қажет. Бұл кезде бұлшықет гликогенінің қоры пайдаланылады, содан кейін — ол жетіспесе — бауыр гликогені. Денедегі гликоген неғұрлым көп болса, оның жұмысы соғұрлым тиімді және ұзағырақ болады. Салыстыратын болсақ, бауырда 100 г гликоген және бұлшықеттерде 400 г сақталады. Мұндай қормен сіз бір күн тамақсыз өмір сүре аласыз.
Гликоген тапшылығы жағдайында не болады?
Гликоген тапшылығы қоректік заттарға толы тағаммен толықтырылуы керек. Спортпен үнемі айналысатын адамдар мұны есте сақтауы керек. Әйтпесе, гликогеннің жетіспеушілігі бұлшықеттердің айтарлықтай әлсіздігіне әкеледі, өйткені олар өздерінің құрылыс блоктары болып табылатын аминқышқылдарынан энергияны ала бастайды. Дегенмен, физикалық жаттығулар жасалды ма, жоқ па, қарамастан, глюкозаның тиісті деңгейін ұстап тұруға бауырдағы гликоген жауап береді.
Дегенмен, физикалық жаттығулар жасалды ма, жоқ па, қарамастан, глюкозаның тиісті деңгейін ұстап тұруға бауырдағы гликоген жауап береді.
Гликоген тапшылығын қалай толтыруға болады?
Тамақты жаттығудан кейін бір сағат ішінде жеген дұрыс. Осы кезеңде ағза берілген көмірсулар мен ақуыздарды барынша тиімді пайдалана алады. Сонымен қатар, майлар көмірсулардың сіңу процесін баяулатады, сондықтан оларды жаттығудан кейін 5 сағат ішінде денеңізге бермеуді ұмытпаңыз. Көмірсулар тамақпен және сұйықтықпен жақсы сіңеді. Жаттығуды аяқтағаннан кейін 6 сағат ішінде шамамен 200 г көмірсуды тұтыну керек. Ең дұрысы, олардың гликемиялық индексі төмен болуы керек. Мұндай өнімдерге басқалар жатады жарма, ботқа, суши, ұзын дәнді күріш, асқабақ, қарбыз, пісірілген бұршақтар.
Содан кейін, 6 сағаттан кейін, қарақұмық, қара бидай наны, манго, киви, шырындар сияқты гликемиялық индексі әлдеқайда төмен өнімдерді жеген дұрыс, бірақ тек қантсыз, пісірілмеген бұршақтар, кебек (сұлы және бидай) және басқалары, оны интернеттен табуға болады. Тек фразаны енгізіңіз: жоғары және төмен гликемиялық индексі бар өнімдер. Осылайша біз нені және қанша жейтінімізге сенімді боламыз және біздің тағамдарымыз пайдалы және теңгерімді болады!
Тек фразаны енгізіңіз: жоғары және төмен гликемиялық индексі бар өнімдер. Осылайша біз нені және қанша жейтінімізге сенімді боламыз және біздің тағамдарымыз пайдалы және теңгерімді болады!
Гликогеннің қызметі
Бауыр гликогені қандағы глюкозаның қалыпты деңгейін сақтауға көмектеседі.
- Жүйке жүйесінің бірқалыпты жұмыс істеуін қолдайды.
- Денедегі қант мөлшері төмендеген кезде глюкоза молекулалары гликогеннен және басқа заттардан қалпына келтіріледі.
- Бұлшықет гликогені бұлшық еттерге қуат береді, соның арқасында біз ұзақ және тиімдірек жаттыға аламыз.
Нарушения обмена гликогена и накопления гликогена
1. Litwack G, Litwack G. Гликоген и гликогенолиз. Биохимия человека. Академическая пресса, 2018: 161-81. [Google Scholar]
2. Роуч П.Дж., Депаоли-роуч А.А., Херли Т.Д. и др.
Гликоген и его метаболизм: некоторые новые разработки и старые темы.
Биохим Дж
2012; 441:763-87. 10.1042/BJ20111416 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
10.1042/BJ20111416 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
3. Тарнопольский М.А. Метаболические миопатии. Континуум (Миннеап Минн) 2016;22:1829-51. 10.1212/CON.0000000000000403 [PubMed] [CrossRef] [Google Scholar]
4. DiMauro S, Spiegel R. Прогресс и проблемы мышечных гликогенозов. Акта Миол 2011;30:96-102. [Бесплатная статья PMC] [PubMed] [Google Scholar]
5. Aynsley-Green A, Williamson DH, Gitzelmann R. Дефицит печеночной гликогенсинтетазы. Определение синдрома по метаболическим и ферментным исследованиям у девочки 9 лет. Арка Дис Чайлд 1977; 52: 573–579. 10.1136/adc.52.7.573 [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
6. Gitzelmann R, Spycher MA, Feil G, et al. Дефицит гликогенсинтазы печени: редко диагностируемое состояние. Eur J Педиатр 1996; 155:561-7. 10.1007/BF01957905 [PubMed] [CrossRef] [Google Scholar]
7. Rutledge SL, Atchison J, Bosshard NU, et al.
Описание клинического случая: дефицит гликогенсинтазы печени — причина кетотической гипогликемии. Педиатрия.
2001;108:495-7. 10.1542/peds.108.2.495 [PubMed] [CrossRef] [Google Scholar]
Педиатрия.
2001;108:495-7. 10.1542/peds.108.2.495 [PubMed] [CrossRef] [Google Scholar]
8. Aynsley-Green A, Williamson DH, Gitzelmann R. Диетическое лечение дефицита гликогенсинтетазы в печени. Хелв Педиатр Акта 1977;32:71-5. [PubMed] [Google Scholar]
9. Aynsley-Green A, Williamson DH, Gitzelmann R. Бессимптомная недостаточность печеночной гликогенсинтетазы. Ланцет 1978; 1:147-8. 10.1016/S0140-6736(78)90442-7 [PubMed] [CrossRef] [Google Scholar]
10. Chen YT, Kishnani PS, Koeberl D. Болезни накопления гликогена. В: Beaudet AL, Vogelstein B, Kinzler KW, et al. редакторы. The Online Metabolic and Molecular Bases of Inherited Disease New York, NY: The McGraw-Hill Companies, Inc., 2014. [Google Scholar]
11. Хеллер С., Ворона Л., Консуэло А. Пищевая терапия болезней накопления гликогена. J Педиатр Гастроэнтерол Нутр 2008;47:S15-21. 10.1097/MPG.0b013e3181818ea5 [PubMed] [CrossRef] [Google Scholar]
12. Orho M, Bosshard NU, Buist NR, et al.
Мутации в гене гликогенсинтазы печени у детей с гипогликемией вследствие болезни накопления гликогена 0 типа. Джей Клин Инвест
1998;102:507-15. 10.1172/JCI2890 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Джей Клин Инвест
1998;102:507-15. 10.1172/JCI2890 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
13. Kasapkara ÇS, Aycan Z, Açoğlu E, et al. Отчет о клиническом случае Вариабельный клинический фенотип трех пациентов с дефицитом печеночной гликогенсинтазы. J Педиатр Эндокринол Метаб 2017;30:459-62. 10.1515/jpem-2016-0317 [PubMed] [CrossRef] [Google Scholar]
14. Smit GP. Отдаленный исход у пациентов с болезнью накопления гликогена Iа типа. Eur J Педиатр 1993;152 Приложение 1:S52-5. 10.1007/BF02072089 [PubMed] [CrossRef] [Google Scholar]
15. Bali DS, Chen YT, Austin S, et al. Болезнь накопления гликогена I типа. GeneReviews®. Сиэтл: Вашингтонский университет, 1993. [Google Scholar]
16. Rake JP, Visser G, Labrune P, et al. Болезнь накопления гликогена I типа: диагностика, лечение, клиническое течение и исход. Результаты европейского исследования болезни накопления гликогена I типа (ESGSD I). Eur J Педиатр 2002;161:S20-34. 10.1007/BF02679990 [PubMed] [CrossRef] [Google Scholar]
17.
18. Wang DQ, Carreras CT, Kishnani PS, et al. Характеристика и патогенез анемии при болезни накопления гликогена типа Ia и Ib. Жене Мед 2012;14:795-9. 10.1038/gim.2012.41 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
19. Сечи А., Дерома Л., Лаполла А. и др. Фертильность и беременность у женщин, страдающих болезнью накопления гликогена I типа, результаты многоцентрового итальянского исследования. J Наследовать Метаб Дис 2013;36:83-9. 10.1007/s10545-012-9490-1 [PubMed] [CrossRef] [Google Scholar]
20. Kishnani PS, Austin SL, Bali DS, et al. Диагностика и лечение болезни накопления гликогена I типа: практическое руководство Американского колледжа медицинской генетики и геномики. Жене Мед 2014;16:e1. 10.1038/gim.2014.128 [PubMed] [CrossRef] [Google Scholar]
21. Trioche P, Francoual J, Capel L, et al.
Полиморфизм аполипопротеина Е и концентрации в сыворотке крови у пациентов с болезнью накопления гликогена типа Ia.
J Наследовать Метаб Дис
2000;23:107-12. 10.1023/A:1005605513534 [PubMed] [CrossRef] [Google Scholar]
Trioche P, Francoual J, Capel L, et al.
Полиморфизм аполипопротеина Е и концентрации в сыворотке крови у пациентов с болезнью накопления гликогена типа Ia.
J Наследовать Метаб Дис
2000;23:107-12. 10.1023/A:1005605513534 [PubMed] [CrossRef] [Google Scholar]
22. Chou JY, Jun HS, Мэнсфилд, Британская Колумбия. Болезни накопления гликогена I типа: нарушения комплексов переносчиков глюкозо-6-фосфатазы/глюкозо-6-фосфата. J Наследовать Метаб Дис 2015;38:511-9. 10.1007/s10545-014-9772-x [PubMed] [CrossRef] [Google Scholar]
23. Qu Y, Abdenur JE, Desnick RJ, et al. Молекулярная пренатальная диагностика болезни накопления гликогена ТИПА Ia. пренат диагностика 1996;16:333-6. 10.1002/(SICI)1097-0223(199604)16:4<333::AID-PD861>3.0.CO;2-G [PubMed] [CrossRef] [Google Scholar]
24. Бхаттачарья К. Диетические дилеммы при лечении болезни накопления гликогена I типа. J Наследовать Метаб Дис 2011;34:621-9. 10.1007/s10545-011-9322-8 [PubMed] [CrossRef] [Google Scholar]
25. Boers SJB, Visser G, Smit PGPA, et al.
Трансплантация печени при болезни накопления гликогена I типа.
Orphanet J Rare Dis
2014;9:47. 10.1186/1750-1172-9-47 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Boers SJB, Visser G, Smit PGPA, et al.
Трансплантация печени при болезни накопления гликогена I типа.
Orphanet J Rare Dis
2014;9:47. 10.1186/1750-1172-9-47 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
26. Chou JY, Jun HS, Мэнсфилд, Британская Колумбия. Болезнь накопления гликогена I типа и дефицит G6Pase-β: этиология и терапия. Нат Рев Эндокринол 2010;6:676-88. 10.1038/nrendo.2010.189 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
27. Gjorgjieva M, Monteillet L, Calderaro J, et al. Поликистоз почек особенности почечной патологии при болезни накопления гликогена I типа: возможная эволюция в почечную неоплазию. J Наследовать Метаб Дис 2018. [Epub перед печатью]. 10.1007/s10545-018-0207-y [PubMed] [CrossRef] [Google Scholar]
28. Lei KJ, Shelly LL, Pan CJ, et al. Мутации в гене глюкозо-6-фосфатазы, вызывающие болезнь накопления гликогена типа 1а. Наука 1993; 262:580-3. 10.1126/science.8211187 [PubMed] [CrossRef] [Google Scholar]
29. Ekstein J, Rubin BY, Weinstein DA, et al.
Частоты мутаций для болезни накопления гликогена Ia у еврейского населения ашкенази.
Am J Med Genet
2004; 129А:162-4. 10.1002/ajmg.a.30232 [PubMed] [CrossRef] [Google Scholar]
Ekstein J, Rubin BY, Weinstein DA, et al.
Частоты мутаций для болезни накопления гликогена Ia у еврейского населения ашкенази.
Am J Med Genet
2004; 129А:162-4. 10.1002/ajmg.a.30232 [PubMed] [CrossRef] [Google Scholar]
30. Visser G, de Jager W, Verhagen LP, et al. На выживание, но не на созревание, влияют предшественники нейтрофилов от пациентов с GSD-1b. J Наследовать Метаб Дис 2012;35:287-300. 10.1007/с10545-011-9379-4 [PubMed] [CrossRef] [Google Scholar]
31. Visser G, Rake J, Labrune P, et al. Гранулоцитарный колониестимулирующий фактор при болезни накопления гликогена типа 1b. Результаты европейского исследования болезни накопления гликогена 1 типа. Eur J Педиатр 2002; 161:S83-7. 10.1007/BF02680001 [PubMed] [CrossRef] [Google Scholar]
32. Melis D, Pivonello R, Parenti G, et al. Повышенная распространенность аутоиммунитета щитовидной железы и гипотиреоза у пациентов с болезнью накопления гликогена I типа. J Pediatr 2007;150:300-5, 305.e1 [PubMed]
33. Hiraiwa H, Pan CJ, Lin B, et al.
Инактивация переносчика глюкозо-6-фосфата вызывает болезнь накопления гликогена типа 1b.
J Биол Хим
1999; 274:5532-6. 10.1074/jbc.274.9.5532 [PubMed] [CrossRef] [Google Scholar]
Hiraiwa H, Pan CJ, Lin B, et al.
Инактивация переносчика глюкозо-6-фосфата вызывает болезнь накопления гликогена типа 1b.
J Биол Хим
1999; 274:5532-6. 10.1074/jbc.274.9.5532 [PubMed] [CrossRef] [Google Scholar]
34. Ercan-Fang N, Gannon MC, Rath VL, et al. Комплексные эффекты множественных модуляторов на гликогенфосфорилазу печени человека а. Am J Physiol Endocrinol Metab 2002;283:E29-37. 10.1152/ajpendo.00425.2001 [PubMed] [CrossRef] [Google Scholar]
35. Кишнани П.С., Чен Ю.Т. Нарушения метаболизма гликогена. В: Клайн М.В. редактор. Rudolph’s Pediatrics, 23e New York, NY: McGraw-Hill Education, 2018. [Google Scholar]
36. Chang S, Rosenberg MJ, Morton H, et al. Выявление мутации гликогенфосфорилазы печени при болезни накопления гликогена VI типа. Хум Мол Жене. 1998;7:865-70. 10.1093/hmg/7.5.865 [PubMed] [CrossRef] [Google Scholar]
37. Burwinkel B, Bakker HD, Herschkovitz E, et al.
Мутации в гене гликогенфосфорилазы печени (PYGL), лежащие в основе гликогеноза типа VI.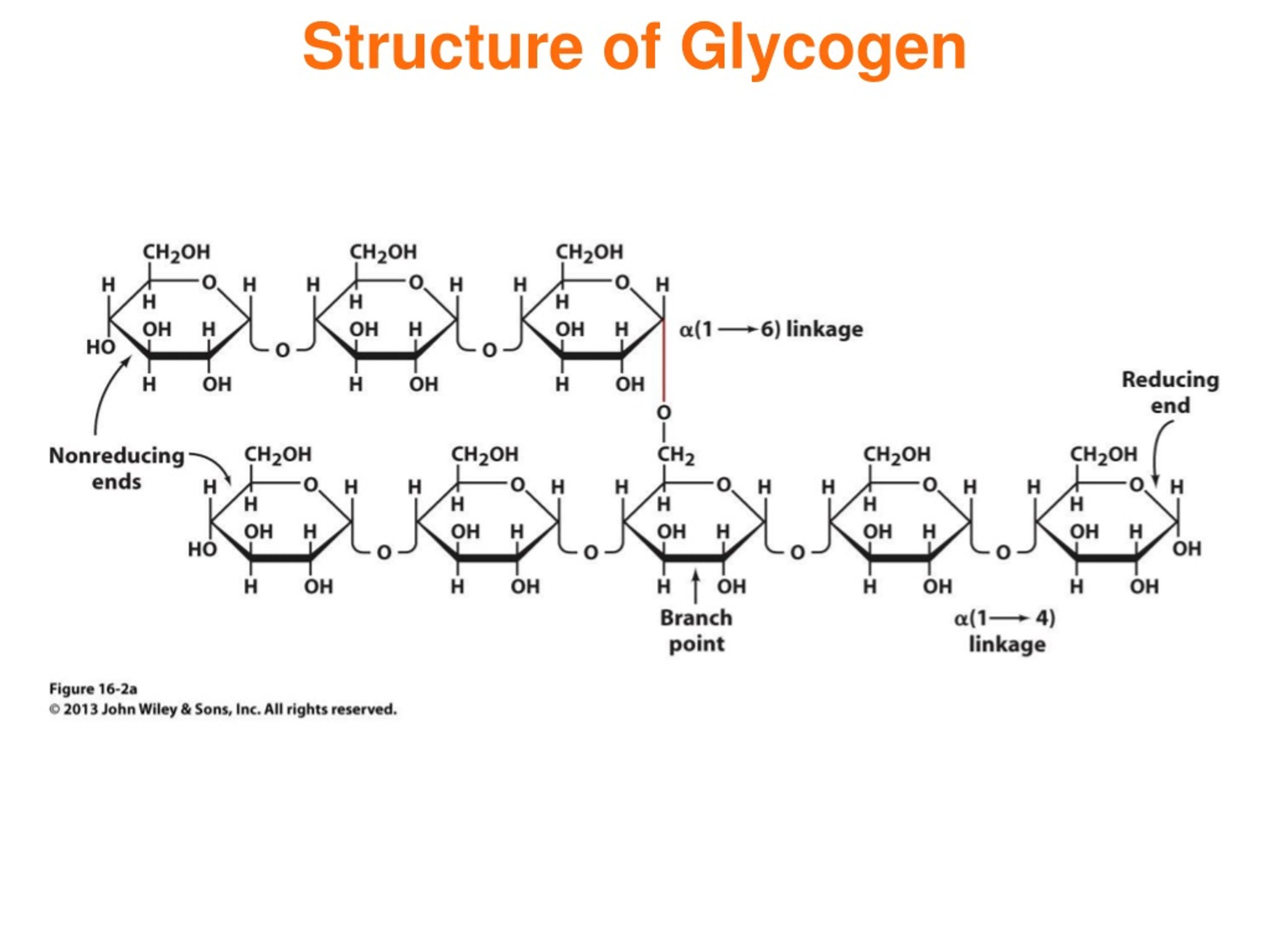 Am J Hum Genet
1998;62:785-91. 10.1086/301790 [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Am J Hum Genet
1998;62:785-91. 10.1086/301790 [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
38. Schimke RN, Zakheim RM, Corder RC, et al. Болезнь накопления гликогена IX типа: доброкачественный гликогеноз печени и недостаточность киназы печеночной фосфорилазы. Дж Педиатр 1973; 83:1031-4. 10.1016/S0022-3476(73)80544-X [PubMed] [CrossRef] [Google Scholar]
39. Beauchamp NJ, Dalton A, Ramaswami U, et al. Болезнь накопления гликогена IX типа: высокая вариабельность клинического фенотипа. Мол Генет Метаб 2007;92:88-99. 10.1016/j.ymgme.2007.06.007 [PubMed] [CrossRef] [Google Scholar]
40. Davidson JJ, Ozçelik T, Hamacher C, et al. Клонирование кДНК печеночной изоформы альфа-субъединицы киназы фосфорилазы и картирование гена в Xp22.2-p22.1, область Х-сцепленного гликогеноза печени человека. Proc Natl Acad Sci U S A 1992;89:2096-100. 10.1073/pnas.89.6.2096 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
41. Burwinkel B, Tanner MS, Kilimann MW. Гликогеноз печени с дефицитом фосфорилазы киназы: прогрессирование до цирроза в младенчестве, связанное с мутациями PHKG2 (h244Y и L225R).
Джей Мед Жене
2000;37:376-7. 10.1136/jmg.37.5.376 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Гликогеноз печени с дефицитом фосфорилазы киназы: прогрессирование до цирроза в младенчестве, связанное с мутациями PHKG2 (h244Y и L225R).
Джей Мед Жене
2000;37:376-7. 10.1136/jmg.37.5.376 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
42. Burwinkel B, Shiomi S, Al Zaben A, et al. Гликогеноз печени из-за дефицита киназы фосфорилазы: структура гена PHKG2 и мутации, связанные с циррозом печени. Хум Мол Жене 1998;7:149-54. 10.1093/hmg/7.1.149 [PubMed] [CrossRef] [Google Scholar]
43. Lucchiari S, Santoro D, Pagliarani S, et al. Клинические, биохимические и генетические особенности недостаточности фермента, расщепляющего гликоген. Акта Миол 2007;26:72-4. [Бесплатная статья PMC] [PubMed] [Google Scholar]
44. Sentner CP, Hoogeveen IJ, Weinstein DA, et al. Болезнь накопления гликогена III типа: диагностика, генотип, лечение, клиническое течение и исход. J Наследовать Метаб Дис 2016;39:697-704. 10.1007/с10545-016-9932-2 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
45. Wolfsdorf JI, Holm IA, Weinstein DA.
Болезни накопления гликогена: фенотипические, генетические и биохимические характеристики и терапия.
Эндокринол Метаб Клин Норт Ам
1999; 28:801-23. 10.1016/S0889-8529(05)70103-1 [PubMed] [CrossRef] [Google Scholar]
Wolfsdorf JI, Holm IA, Weinstein DA.
Болезни накопления гликогена: фенотипические, генетические и биохимические характеристики и терапия.
Эндокринол Метаб Клин Норт Ам
1999; 28:801-23. 10.1016/S0889-8529(05)70103-1 [PubMed] [CrossRef] [Google Scholar]
46. Kishnani PS, Austin SL, Arn P, et al. Рекомендации по диагностике и лечению болезни накопления гликогена III типа. Жене Мед 2010;12:446-63. 10.1097/GIM.0b013e3181e655b6 [PubMed] [CrossRef] [Google Scholar]
47. Mayorandan S, Meyer U, Das AM, et al. Болезнь накопления гликогена III типа: модифицированная диета Аткинса улучшает миопатию. Orphanet J Rare Dis 2014;9:196. 10.1186/s13023-014-0196-3 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
48. Bruno C, van Diggelen OP, Cassandrini D, et al. Клинико-генетическая гетерогенность дефицита ферментов ветвления (гликогеноз IV типа). неврология 2004;63:1053-8. 10.1212/01.WNL.0000138429.11433.0D [PubMed] [CrossRef] [Google Scholar]
49. Burrow TA, Hopkin RJ, Bove KE, et al. Нелетальная врожденная гипотония вследствие болезни накопления гликогена IV типа.
Am J Med Genet Часть A
2006; 140:878-82. 10.1002/ajmg.a.31166 [PubMed] [CrossRef] [Google Scholar]
Нелетальная врожденная гипотония вследствие болезни накопления гликогена IV типа.
Am J Med Genet Часть A
2006; 140:878-82. 10.1002/ajmg.a.31166 [PubMed] [CrossRef] [Google Scholar]
50. Bruno C, Cassandrini D, Di Mauro S, et al. Нервно-мышечные формы дефицита фермента ветвления гликогена. Акта Миол 2007;26:75-8. [Бесплатная статья PMC] [PubMed] [Google Scholar]
51. Szymanska E, Szymanska S, Truszkowska G, et al. Вариабельная клиническая картина болезни накопления гликогена IV типа: от выраженной гепатоспленомегалии до сердечной недостаточности. Некоторые расхождения в генетических и биохимических аномалиях. Arch Med Sci 2018;14:237-47. 10.5114/aoms.2018.72246 [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
52. Greene HL, Brown BI, McClenathan DT, et al. Новый вариант гликогеноза IV типа: дефицит активности ферментов ветвления без выраженного прогрессирующего заболевания печени. гепатология 1988;8:302-6. 10.1002/hep.1840080219 [PubMed] [CrossRef] [Google Scholar]
53. McConkie-Rosell A, Wilson C, Kishnani P, et al.
Клинические и лабораторные данные у четырех пациентов с непрогрессирующей печеночной формой болезни накопления гликогена IV типа.
J Наследовать Метаб Дис
1996;19:51-8. 10.1007/BF01799348 [PubMed] [CrossRef] [Google Scholar]
McConkie-Rosell A, Wilson C, Kishnani P, et al.
Клинические и лабораторные данные у четырех пациентов с непрогрессирующей печеночной формой болезни накопления гликогена IV типа.
J Наследовать Метаб Дис
1996;19:51-8. 10.1007/BF01799348 [PubMed] [CrossRef] [Google Scholar]
54. Roscher A, Patel J, Hewson S, et al. Естественная история болезни накопления гликогена типов VI и IX: долгосрочный результат из крупнейшего метаболического центра в Канаде. Мол Генет Метаб 2014;113:171-6. 10.1016/j.ymgme.2014.09.005 [PubMed] [CrossRef] [Google Scholar]
55. Kollberg G, Tulinius M, Gilljam T, et al. Кардиомиопатия и непереносимость физической нагрузки при болезни накопления мышечного гликогена 0. N Engl J Med 2007; 357:1507-14. 10.1056/NEJMoa066691 [PubMed] [CrossRef] [Google Scholar]
56. Cameron JM, Levandovskiy V, MacKay N, et al.
Идентификация новой мутации в GYS1 (специфическая для мышц гликогенсинтаза), приводящей к внезапной сердечной смерти, которую можно диагностировать по кожным фибробластам.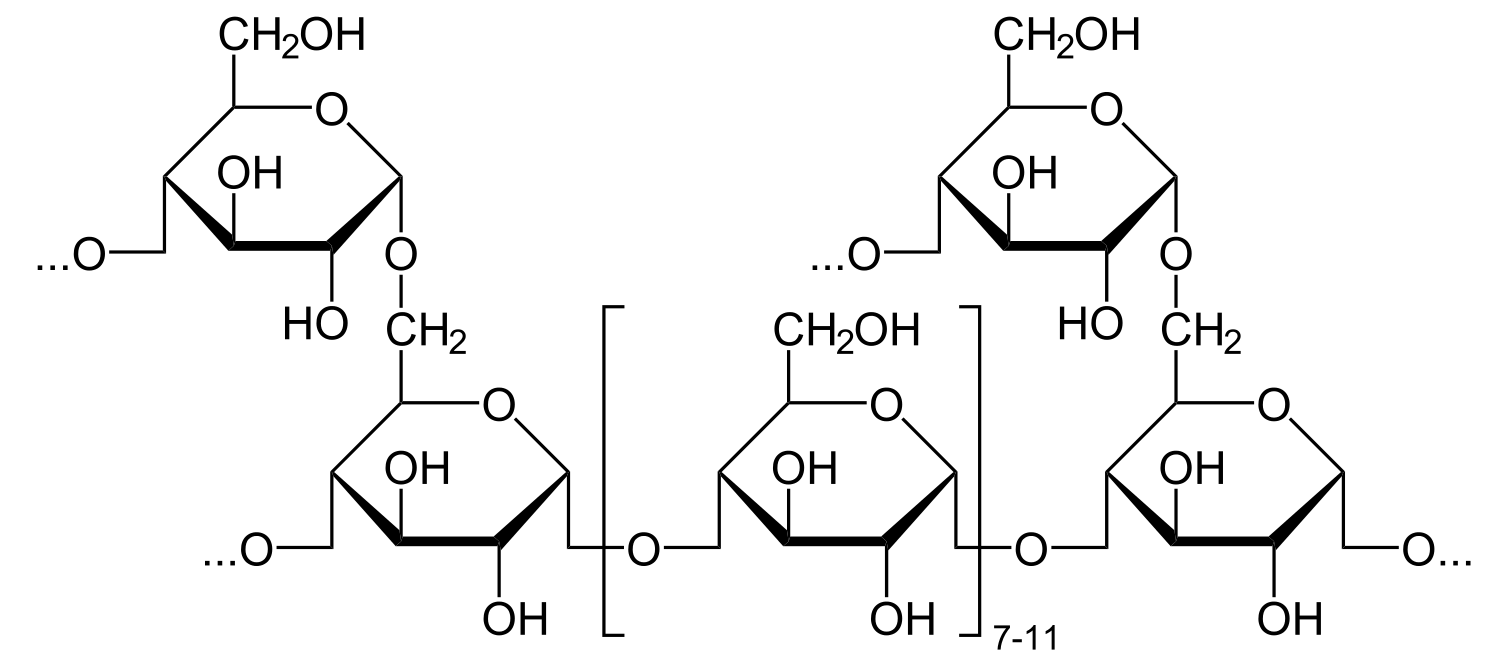 Мол Генет Метаб
2009;98:378-82. 10.1016/j.ymgme.2009.07.012 [PubMed] [CrossRef] [Google Scholar]
Мол Генет Метаб
2009;98:378-82. 10.1016/j.ymgme.2009.07.012 [PubMed] [CrossRef] [Google Scholar]
57. Sukigara S, Liang WC, Komaki H, et al. Болезнь накопления мышечного гликогена 0, проявляющаяся периодическими обмороками со слабостью и миалгией. Нервно-мышечное расстройство 2012;22:162-5. 10.1016/j.nmd.2011.08.008 [PubMed] [CrossRef] [Google Scholar]
58. Лим Дж. А., Ли Л., Рабен Н. Болезнь Помпе: от патофизиологии к терапии и обратно. Фронт старения Neurosci 2014;6:177. 10.3389/fnagi.2014.00177 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
59. Слоним А.Е., Булоне Л., Ритц С. и соавт. Идентификация двух подтипов дефицита кислой мальтазы у детей. Дж Педиатр 2000;137:283-5. 10.1067/mpd.2000.107112 [PubMed] [CrossRef] [Google Scholar]
60. Güngör D, Reuser AJJ. Как описать клинический спектр болезни Помпе? Am J Med Genet Часть A 2013;161А:399-400. 10.1002/ajmg.a.35662 [PubMed] [CrossRef] [Google Scholar]
61. Amartino H, Painceira D, Pomponio R, et al.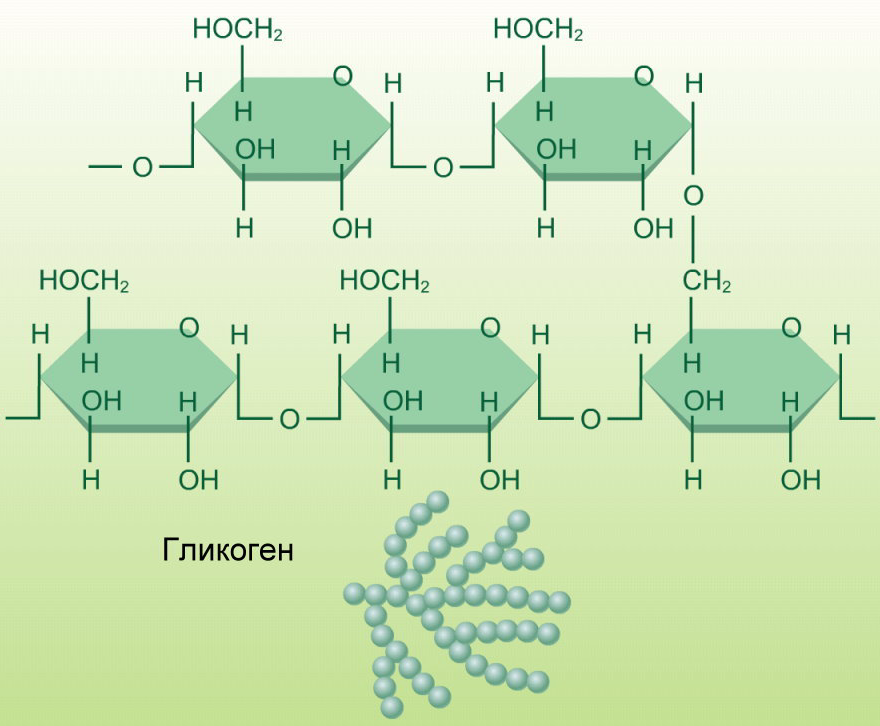 Две клинические формы болезни накопления гликогена II типа в двух поколениях одной семьи.
Клин Жене
2006;69:187-8. 10.1111/j.1399-0004.2005.00557.x [PubMed] [CrossRef] [Google Scholar]
Две клинические формы болезни накопления гликогена II типа в двух поколениях одной семьи.
Клин Жене
2006;69:187-8. 10.1111/j.1399-0004.2005.00557.x [PubMed] [CrossRef] [Google Scholar]
62. Kishnani PS, Beckemeyer AA, Mendelsohn NJ. Новая эра болезни Помпе: достижения в обнаружении, понимании фенотипического спектра, патофизиологии и лечении. Am J Med Genet Часть C Semin Med Genet 2012;160С:1-7. 10.1002/ajmg.c.31324 [PubMed] [CrossRef] [Google Scholar]
63. Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, et al. Корреляция генотип-фенотип при болезни Помпе. Am J Med Genet Часть C Semin Med Genet 2012;160С:59-68. 10.1002/ajmg.c.31318 [PubMed] [CrossRef] [Google Scholar]
64. Bembi B, Cerini E, Danesino C, et al. Диагностика гликогеноза II типа. неврология 2008;71:S4-11. 10.1212/WNL.0b013e31818da91e [PubMed] [CrossRef] [Google Scholar]
65. Chien YH, Hwu WL, Lee NC.
Болезнь Помпе: Ранняя диагностика и раннее лечение имеют значение.
Педиатр Неонатол
2013;54:219-27.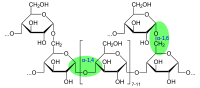 10.1016/j.pedneo.2013.03.009 [PubMed] [CrossRef] [Google Scholar]
10.1016/j.pedneo.2013.03.009 [PubMed] [CrossRef] [Google Scholar]
66. Bodamer OA, Scott CR, Giugliani R. Скрининг новорожденных на болезнь Помпе. Педиатрия 2017;140:S4-13. 10.1542/peds.2016-0280C [PubMed] [CrossRef] [Google Scholar]
67. Banugaria SG, Prater SN, Ng YK, et al. Влияние антител на клинические исходы заболеваний, леченных терапевтическим белком: уроки, извлеченные из детской болезни Помпе. Жене Мед 2011;13:729-36. 10.1097/GIM.0b013e3182174703 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
68. van der Ploeg AT, Clemens PR, Corzo D, et al. Рандомизированное исследование алглюкозидазы альфа при болезни Помпе с поздним началом. N Engl J Med 2010; 362:1396-406. 10.1056/NEJMoa0909859 [PubMed] [CrossRef] [Google Scholar]
69. Haller RG, Wyrick P, Taivassalo T, et al. Аэробное кондиционирование: эффективное лечение болезни Макардла. Энн Нейрол 2006;59:922-8. 10.1002/ana.20881 [PubMed] [CrossRef] [Google Scholar]
70. Виссинг Дж. , Халлер Р.Г.
Влияние пероральной сахарозы на толерантность к физической нагрузке у пациентов с болезнью Макардла.
N Engl J Med
2003;349:2503-9. 10.1056/NEJMoa031836 [PubMed] [CrossRef] [Google Scholar]
, Халлер Р.Г.
Влияние пероральной сахарозы на толерантность к физической нагрузке у пациентов с болезнью Макардла.
N Engl J Med
2003;349:2503-9. 10.1056/NEJMoa031836 [PubMed] [CrossRef] [Google Scholar]
71. Haller RG. Лечение болезни Мак-Ардла. Арка Нейрол 2000;57:923-4. 10.1001/archneur.57.7.923 [PubMed] [CrossRef] [Google Scholar]
72. Nakajima H, Raben N, Hamaguchi T, et al. Дефицит фосфофруктокиназы; прошлое, настоящее и будущее. Курр Мол Мед 2002;2:197-212. 10.2174/1566524024605734 [PubMed] [CrossRef] [Google Scholar]
73. Тоскано А., Мусумечи О. Болезнь Таруи и дистальные гликогенозы: клиническое и генетическое обновление. Акта Миол 2007;26:105-7. [Бесплатная статья PMC] [PubMed] [Google Scholar]
74. Preisler N, Orngreen MC, Echaniz-Laguna A, et al. Дефицит киназы мышечной фосфорилазы: нейтральный метаболический вариант или болезнь? неврология 2012;78:265-8. 10.1212/WNL.0b013e31824365f9 [PubMed] [CrossRef] [Google Scholar]
75. Vissing J, Quistorff B, Haller RG. Влияние топлива на толерантность к физической нагрузке при дефиците фосфоглицератмутазы в мышцах.
Арка Нейрол
2005;62:1440-3. 10.1001/archneur.62.9.1440 [PubMed] [CrossRef] [Google Scholar]
Влияние топлива на толерантность к физической нагрузке при дефиците фосфоглицератмутазы в мышцах.
Арка Нейрол
2005;62:1440-3. 10.1001/archneur.62.9.1440 [PubMed] [CrossRef] [Google Scholar]
76. Ку Б., Оскарссон Б. Дефицит фосфоглицератмутазы (болезнь накопления гликогена X), вызванный новым вариантом PGAM-M. Нервно-мышечное расстройство 2016;26:688-90. 10.1016/j.nmd.2016.08.002 [PubMed] [CrossRef] [Google Scholar]
77. Naini A, Toscano A, Musumeci O, et al. Еще раз о дефиците фосфоглицератмутазы в мышцах. Арка Нейрол 2009;66:394-8. 10.1001/archneurol.2008.584 [PubMed] [CrossRef] [Google Scholar]
78. Запись OMIM — № 612933 — БОЛЕЗНЬ НАКОПЛЕНИЯ ГЛИКОГЕНА XI; GSD11 [цитировано 9 октября 2018 г.]. Доступно в Интернете: https://www.omim.org/entry/612933?search=gsd11&highlight=gsd11
79. Anai T, Urata K, Tanaka Y, et al.
Беременность, осложненная дефицитом М-субъединицы лактатдегидрогеназы: описание первого случая.
J Obstet Gynaecol Res
2002; 28:108-11. 10.1046/j. 1341-8076.2002.00015.x [PubMed] [CrossRef] [Google Scholar]
1341-8076.2002.00015.x [PubMed] [CrossRef] [Google Scholar]
80. Wakabayashi H, Tsuchiya M, Yoshino K, et al. Наследственный дефицит Н-субъединицы лактатдегидрогеназы. Интерн Мед 1996;35:550-4. 10.2169/internalmedicine.35.550 [PubMed] [CrossRef] [Google Scholar]
81. Santer R, Groth S, Kinner M, et al. Спектр мутаций гена облегчающего переносчика глюкозы SLC2A2 (GLUT2) у пациентов с синдромом Фанкони-Биккеля. Хум Жене 2002;110:21-9. 10.1007/s00439-001-0638-6 [PubMed] [CrossRef] [Google Scholar]
82. Mamoune A, Bahuau M, Hamel Y, et al. Термолабильная мутантная альдолаза А вызывает лихорадочный рецидивирующий рабдомиолиз без гемолитической анемии. Уилки АОМ, редактор. PLoS Genet 2014;10:e1004711. [Бесплатная статья PMC] [PubMed] [Google Scholar]
83. Du S, Guan Z, Hao L, et al.
Альдолаза фруктозо-бисфосфата а является потенциальным ассоциированным с метастазами маркером плоскоклеточной карциномы легкого и способствует онкогенезу и миграции клеток легкого.
PLoS Один
2014;9:e85804. 10.1371/journal.pone.0085804 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
10.1371/journal.pone.0085804 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
84. Comi GP, Fortunato F, Lucchiari S, et al. Дефицит бета-энолазы, новая метаболическая миопатия дистального гликолиза. Энн Нейрол 2001;50:202-7. 10.1002/ana.1095 [PubMed] [CrossRef] [Google Scholar]
85. Musumeci O, Brady S, Rodolico C, et al. Рецидивирующий рабдомиолиз из-за дефицита мышечной β-энолазы: очень редко или недооценен? Джей Нейрол 2014; 261:2424-8. 10.1007/s00415-014-7512-7 [PubMed] [CrossRef] [Google Scholar]
86. Морава Э., Вонг С., Лефебер Д. Тяжесть заболевания и клинический исход при недостаточности фосфоглюкомутазы. J Наследовать Метаб Дис 2015;38:207-9. 10.1007/s10545-014-9769-5 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
87. Wong SY, Gadomski T, Kozicz T, et al. Определение фенотипа и оценка тяжести дефицита фосфоглюкомутазы-1. Дж Педиатр 2016;175:130-136.e8. 10.1016/j.jpeds.2016.04.021 [PubMed] [CrossRef] [Google Scholar]
88. Hedberg-Oldfors C, Glamuzina E, Ruygrok P, et al.
Кардиомиопатия как признак дефицита гликогенина-1 — отчет о трех случаях и обзор литературы.
J Наследовать Метаб Дис
2017;40:139-49. 10.1007/s10545-016-9978-1 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Hedberg-Oldfors C, Glamuzina E, Ruygrok P, et al.
Кардиомиопатия как признак дефицита гликогенина-1 — отчет о трех случаях и обзор литературы.
J Наследовать Метаб Дис
2017;40:139-49. 10.1007/s10545-016-9978-1 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
89. Stemmerik MG, Madsen KL, Laforet P, et al. Синтез и расщепление мышечного гликогена нарушаются при дефиците гликогенина-1. неврология 2017;89:2491-4. 10.1212/WNL.0000000000004752 [PubMed] [CrossRef] [Google Scholar]
90. Danon MJ, Oh SJ, DiMauro S, et al. Лизосомальная болезнь накопления гликогена с нормальной кислой мальтазой. неврология 1981; 31:51-7. 10.1212/WNL.31.1.51 [PubMed] [CrossRef] [Google Scholar]
91. Schorderet DF, Cottet S, Lobrinus JA, et al. Ретинопатия при болезни Данона. Арка Офтальмол 2007;125:231-6. 10.1001/archopht.125.2.231 [PubMed] [CrossRef] [Google Scholar]
92. Majer F, Piherova L, Reboun M, et al.
Вариации числа экзонов-копий LAMP2 у женщин-пробандов, гетерозиготных по болезни Данон: нечасто или недостаточно выявлено?
Am J Med Genet Часть A
2018;176:2430-4.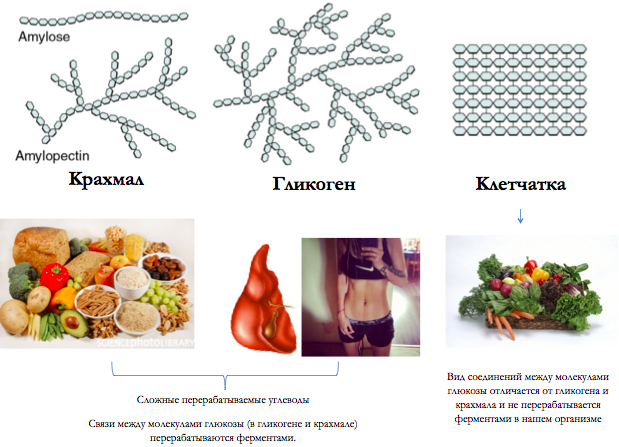 10.1002/ajmg.a.40430 [PubMed] [CrossRef] [Google Scholar]
10.1002/ajmg.a.40430 [PubMed] [CrossRef] [Google Scholar]
93. Burwinkel B, Scott JW, Bührer C, et al. Фатальный врожденный гликогеноз сердца, вызванный рецидивирующей активирующей мутацией R531Q в гамма-2-субъединице АМФ-активируемой протеинкиназы (PRKAG2), а не дефицитом киназы фосфорилазы. Am J Hum Genet 2005;76:1034-49. 10.1086/430840 [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
94. Ramachandran N, Girard JM, Turnbull J, et al. Аутосомно-рецессивно наследуемые прогрессирующие миоклонус-эпилепсии и их гены. Эпилепсия 2009 г.;50:29-36. 10.1111/j.1528-1167.2009.02117.x [PubMed] [CrossRef] [Google Scholar]
Химическая логика… синтеза и деградации гликогена
Химическая логика… синтеза и деградации гликогенаХимическая логика… синтеза и расщепления гликогена
проф. Доутор Педро Силва
Адъюнкт-профессор Университета Фернандо Пессоа
| Другие метаболические пути: | Метаболизм жирных кислот Гликолиз Цикл лимонной кислоты Ферментация и дыхание Синтез гликогена и гликогенолиз Глюконеогенез Деградация аминокислот и цикл мочевины Пентозофосфатный путь Метаболическая регуляция и интеграция Анимации по метаболической и структурной биохимии |
Уровни глюкозы в крови поддерживаются приблизительно на постоянном уровне около 4-5 мМ. Глюкоза проникает в клетки путем облегченной диффузии. Поскольку этот процесс не позволяет клетке содержать глюкозу в более высокой концентрации, чем та, которая присутствует в кровотоке, клетка (посредством энксимы гексокиназы) химически модифицирует глюкозу путем фосфорилирования:
Глюкоза проникает в клетки путем облегченной диффузии. Поскольку этот процесс не позволяет клетке содержать глюкозу в более высокой концентрации, чем та, которая присутствует в кровотоке, клетка (посредством энксимы гексокиназы) химически модифицирует глюкозу путем фосфорилирования:
Поскольку клеточная мембрана непроницаема для глюкозо-6-фосфата, этот процесс эффективно «улавливает» глюкозу внутри клетки, позволяя извлекать большее количество глюкозы из кровотока. Глюкозо-6-фосфат будет использоваться в синтезе гликогена. (форма хранения глюкозы), образование других соединений углерода по пентозофосфатному пути или расщепление с целью получения энергии.0195 гликолиз .
Большое количество глюкозы-6-Ф внутри клетки вызывает повышение осмотического давления. В этих условиях вода будет стремиться попасть в клетку, увеличивая ее колум и (в конечном счете) лизируя ее. Чтобы предотвратить это, клетка запасает глюкозу-6-Ф.
в виде полимера: гликогена .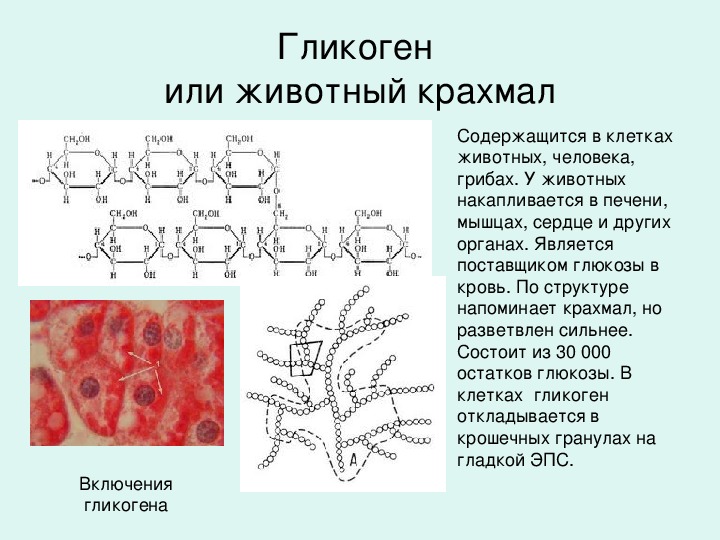 Гликоген представляет собой малорастворимый (и поэтому осмотически неактивный) разветвленный полисахарид, состоящий из мономеров глюкозы, соединенных гликозидными связями типа а-1,4 и а-1,6.
(в точках разветвления):
Гликоген представляет собой малорастворимый (и поэтому осмотически неактивный) разветвленный полисахарид, состоящий из мономеров глюкозы, соединенных гликозидными связями типа а-1,4 и а-1,6.
(в точках разветвления):
Для использования в синтезе гликогена глюкоза-6-фосфат сначала изомеризуется в глюкозо-1-фосфато ферментом фосфоглюкомутазой .
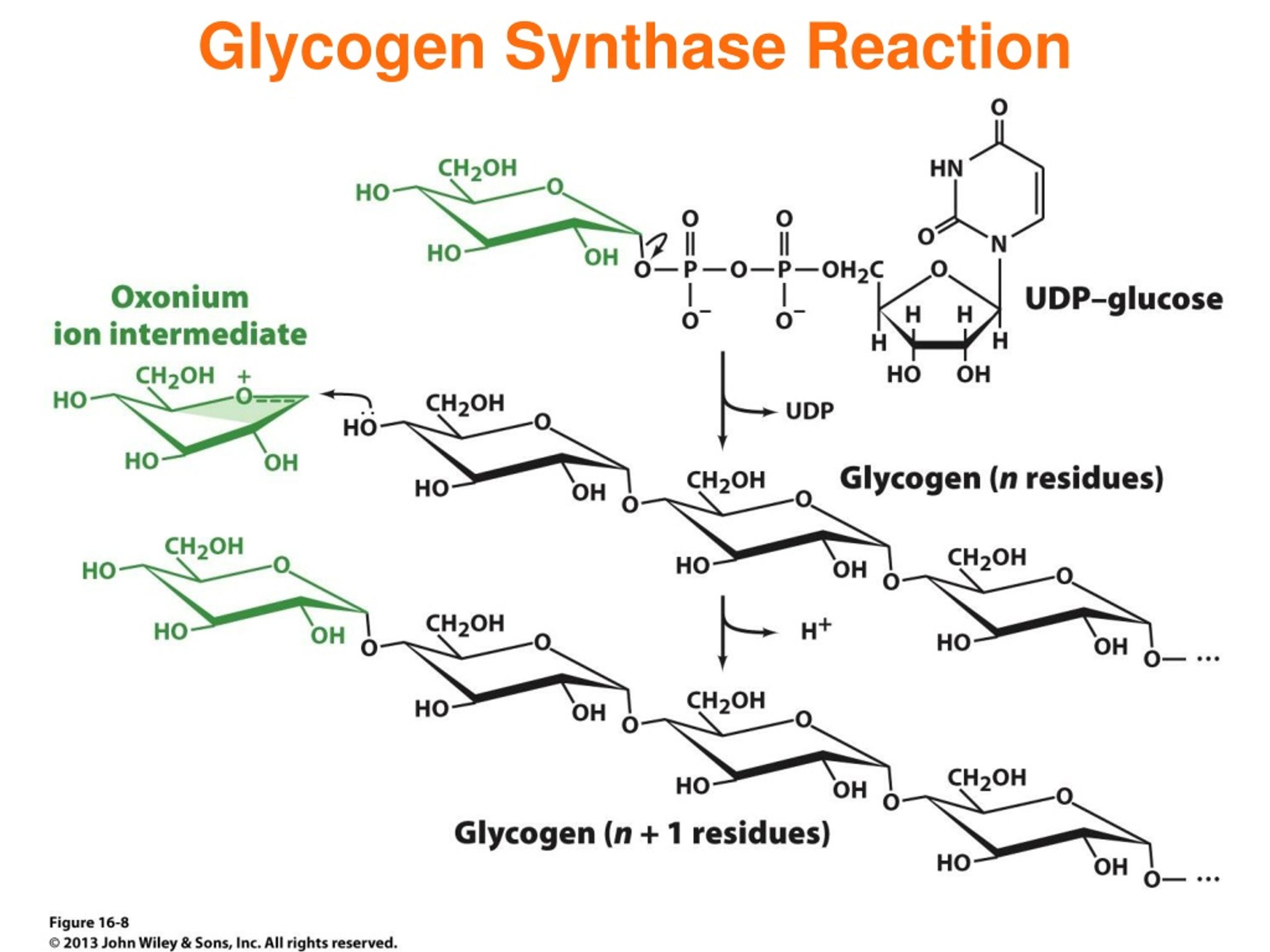
Присоединение глюкозы-1-P к 4′-углероду цепи гликогена термодинамически невыгодно, поскольку потенциал переноса фосфата связей C-O-P довольно низок. Таким образом, глюкоза-1-P будет активирована , т. е. превратились в вид с высоким потенциалом переноса фосфатов. Это достигается реакцией с трифосфатом уридина (UTP, аналог АТФ, с заменой аденина уридином).
Сама по себе эта реакция не является термодинамически благоприятной. Однако пирофосфат ( PPi ), высвобождаемый в этой реакции, может гидролизоваться вездесущим ферментом пирофосфатазой в очень экзергонической реакции. Удаление PPi смещает равновесие в сторону образования УДФ-глюкозы, что иллюстрирует общий принцип, заключающийся в том, что очень экзэргоническая реакция может быть соединена с неблагоприятной в других отношениях реакцией, чтобы сделать ее спонтанной.
Удаление PPi смещает равновесие в сторону образования УДФ-глюкозы, что иллюстрирует общий принцип, заключающийся в том, что очень экзэргоническая реакция может быть соединена с неблагоприятной в других отношениях реакцией, чтобы сделать ее спонтанной.
УДФ-глюкоза обладает высоким потенциалом переноса фосфатов, что позволяет ей отдавать глюкозу на 4′-конец гликогеновой цепи в реакции, катализируемой гликогенсинтазой:
Гликогенсинтаза может присоединять глюкозу только к уже существующим цепям гликогена, т. е. она не может начать синтез новой молекулы гликогена. Синтез гликогена начинается с присоединения молекулы глюкозы к остатку тирозина, находящемуся в активном центре белка, называемого 9.0195 гликогенин . После добавления примерно семи молекул глюкозы новая цепь гликогена готова к действию гликогенсинтазы.
Точки ветвления создаются «ферментом ветвления». Этот фермент действует на линейные участки гликогена, содержащие не менее 11 молекул глюкозы. Ветвящийся фермент
(амило(1,4 —> 1,6)-трансгликозилаза) переносит концевые сегменты гликогена длиной 7 молекул глюкозы на ОН-группу углерода 6 остатка глюкозы (в той же или в другой цепи). Точки ветвления должны находиться на расстоянии не менее 4 молекул глюкозы друг от друга.
Ветвящийся фермент
(амило(1,4 —> 1,6)-трансгликозилаза) переносит концевые сегменты гликогена длиной 7 молекул глюкозы на ОН-группу углерода 6 остатка глюкозы (в той же или в другой цепи). Точки ветвления должны находиться на расстоянии не менее 4 молекул глюкозы друг от друга.
Гликоген расщепляется последовательным действием трех ферментов:
- гликогенфосфорилаза расщепляет(1-4) связывается с неорганическим фосфатом (Pi). Он может расщеплять остатки глюкозы только на расстоянии 4 (или более) остатков глюкозы от точки ветвления. Он использует пиридоксаль, производное витамина B 6 , в качестве кофактора.
Молекула гликогена с ответвлениями только из четырех молекул глюкозы («предельный декстрин») не может быть далее расщеплена только гликогенфосфорилазой. Ему нужен другой фермент:
- фермент, разветвляющий гликоген :
переносит три остатка глюкозы с предельной ветви на другую.
 Последний остаток в разветвлении (с гликозидной связью (1-6) ) удаляется 90–195 гидролизом 90–196 с образованием свободной глюкозы и разветвленного гликогена. Гидролиз этого остатка катализируется тем же расщепляющим ферментом.
Последний остаток в разветвлении (с гликозидной связью (1-6) ) удаляется 90–195 гидролизом 90–196 с образованием свободной глюкозы и разветвленного гликогена. Гидролиз этого остатка катализируется тем же расщепляющим ферментом.Гликогенфосфорилаза действует намного быстрее, чем деветвящий фермент, и поэтому внешние ветви гликогена очень быстро расщепляются в мышцах, когда требуется много энергии. Расщепление гликогена после этой точки требует действия деветвящего фермента и, следовательно, происходит медленнее, что частично объясняет тот факт, что мышца может выполнять максимальное напряжение только в течение нескольких секунд.
- фосфоглюкомутаза : катализирует изомеризацию глюкозо-1-Ф в глюкозу-6-Ф, и наоборот:
Глюкозо-6-фосфат можно использовать в гликолизе. В отличие от мышц, печень (и, в меньшей степени, почки) содержит глюкозо-6-фосфатазу, гидролитический фермент, катализирующий дефосфорилатон глюкозо-6-фосфата, что позволяет ей поставлять глюкозу в другие ткани:
| Биохимия,
Дональд Воет и Джудит Воет Отличный текст. Вам так же может понравиться... |

