1.2 Этиология и патогенез \ КонсультантПлюс
1.2 Этиология и патогенез
Причиной возникновения ГБ Ia типа являются мутации в гене G6PC, кодирующем глюкозо-6-фосфатазу, что приводит к ее недостаточности в печени, почках, слизистой оболочке кишечника, а также в островках поджелудочной железы и желчного пузыря. Генный локус гликогеновой болезни Ia типа соответствует 17q21.31. Он содержит 5 экзонов и занимает около 12,5 kb геномной ДНК. Тип наследования — аутосомно-рецессивный.
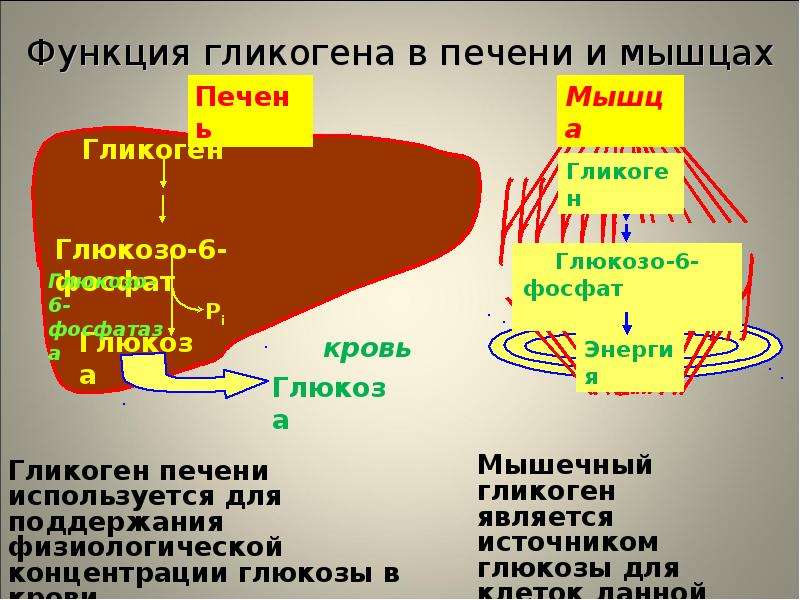
Патогенез ГБ Ia типа. Глюкозо-6-фосфатаза катализирует конечную реакцию как глюконеогенеза, так и гидролиза гликогена, гидролизуя глюкозо-6-фосфат на глюкозу и неорганический фосфат (Pi) и являясь единственным источником обеспечения организма большими концентрациями глюкозы. Неспособность организма больного превратить глюкозо-6-фосфат в глюкозу ведет к гипогликемии даже при кратковременном голодании из-за блокады гликогенолиза и глюконеогенеза и к накоплению гликогена в печени, почках и слизистой оболочке кишечника, приводя к дисфункции этих органов. Накопление субстрата блокированной реакции, глюкозо-6-фосфата, стимулирует гликолиз и накопление лактата, который, синтезируясь в эритроцитах и мышечной ткани, не может быть превращен в глюкозу в печени (блокада глюконеогенеза). Гипогликемия обусловливает относительно низкую концентрацию инсулина. Снижается соотношение инсулин/глюкагон, стимулируя липолиз, и повышается уровень жирных кислот в плазме. Стимуляция гликолиза ведет к увеличению синтеза глицерола и ацетил-КоА, субстратов и кофакторов синтеза триглицеридов в печени. Глюкагон также стимулирует метаболические пути, угнетающие жирных кислот в митохондриях, что сопровождается дикарбоновой ацидурией. Если скорость синтеза аполипопротеинов и образования липопротеинов очень низкой плотности (ЛПОНП) и липопротеинов низкой плотности (ЛПНП) отстает от ускоренного синтеза липидов, в гепатоцитах образуются капли жира, вызывая выраженную гепатомегалию (жировая трансформация, стеатоз печени). Лактат является конкурентным ингибитором почечно-канальцевой секреции уратов, что ведет к гиперурикемии и гипоурикозурии.
Накопление субстрата блокированной реакции, глюкозо-6-фосфата, стимулирует гликолиз и накопление лактата, который, синтезируясь в эритроцитах и мышечной ткани, не может быть превращен в глюкозу в печени (блокада глюконеогенеза). Гипогликемия обусловливает относительно низкую концентрацию инсулина. Снижается соотношение инсулин/глюкагон, стимулируя липолиз, и повышается уровень жирных кислот в плазме. Стимуляция гликолиза ведет к увеличению синтеза глицерола и ацетил-КоА, субстратов и кофакторов синтеза триглицеридов в печени. Глюкагон также стимулирует метаболические пути, угнетающие жирных кислот в митохондриях, что сопровождается дикарбоновой ацидурией. Если скорость синтеза аполипопротеинов и образования липопротеинов очень низкой плотности (ЛПОНП) и липопротеинов низкой плотности (ЛПНП) отстает от ускоренного синтеза липидов, в гепатоцитах образуются капли жира, вызывая выраженную гепатомегалию (жировая трансформация, стеатоз печени). Лактат является конкурентным ингибитором почечно-канальцевой секреции уратов, что ведет к гиперурикемии и гипоурикозурии.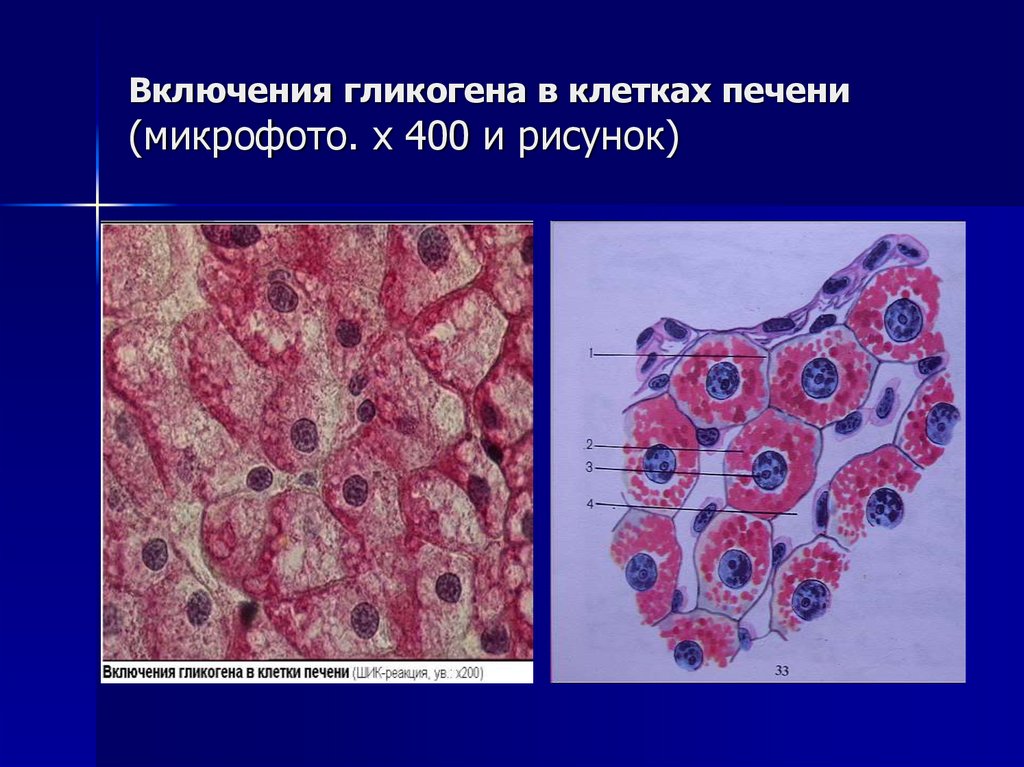 Гиперурикемия является следствием не только снижения почечного клиренса, но и гиперпродукции мочевой кислоты в результате истощения внутрипеченочного фосфата и ускоренной деградации адеиновых нуклеотидов.
Гиперурикемия является следствием не только снижения почечного клиренса, но и гиперпродукции мочевой кислоты в результате истощения внутрипеченочного фосфата и ускоренной деградации адеиновых нуклеотидов.
Механизмы формирования фокально-сегментарного гломерулосклероза при ГБ I типа окончательно не ясны. Не может быть причиной гиперфильтации и характер питания, т.к. в рационе преобладают углеводы. Не исключено, что формирование фокально-сегментарного гломерулосклероза связано также с персистирующей гиперлипидемией, наподобие того, как это происходит при первичном нефротическом синдроме с неадекватным контролем частоты рецидивов и активности болезни. На роль других патогенетических факторов претендуют артериальная гипертензия, нередко возникающая при ГБ I типа, и гиперурикемия. Конкременты чаще состоят из кристаллов моногидрата оксалата кальция, хотя наличие гиперуратурии дает основания ожидать уратного состава камней, но это не так. Механизм образования оксалатно-кальциевых конкрементов при ГБ I типа не изучен. Возможно, к нему имеют отношение хронический ацидоз и урикемия либо энзиматические дефекты в печени с возможной аналогией гипероксалурии типа I.
Возможно, к нему имеют отношение хронический ацидоз и урикемия либо энзиматические дефекты в печени с возможной аналогией гипероксалурии типа I.
Причиной возникновения ГБ Ib типа являются мутации гена SLC17A4, кодирующего микросомальный транспортный белок T1 (транслоказу глюкозо-6-фосфатазы), что приводит к ее недостаточности в печени, почках, слизистой оболочке кишечника. Генный локус ГБ Ib соответствует 11q23.3. Он содержит 9 экзонов и занимает от 4 до 5,3 kb геномной ДНК. Тип наследования — аутосомно-рецессивный.
Патогенез ГБ Ib типа. Расположение активного центра глюкозо-6-фосфатазы в просвете ЭПР обусловливает необходимость транспортировки всех субстратов и продуктов реакции, катализируемой ферментом, через мембрану ЭПР. Недостаточность белка T1 сопровождается недостаточностью глюкозо-6-фосфатазы. Специфическим признаком ГБ Ib типа является нейтропения и ухудшение функции нейтрофилов. У таких больных нарушена как двигательная способность нейтрофилов, так и их активность при респираторном взрыве. Кроме того, эти патологические изменения могут быть связаны с нарушением транспорта глюкозы через мембрану полиморфно-ядерных лейкоцитов. Согласно существующим предположениям, микросомальный транспорт глюкозо-6-фосфата играет определенную роль в антиоксидантной защите нейтрофилов, а генетическая поломка транслоказы глюкозо-6-фосфатазы ведет к нарушению клеточных функций, в частности, апоптоза, что может служить объяснением нейтрофильной дисфункции у больных с ГБ Ib типа.
Кроме того, эти патологические изменения могут быть связаны с нарушением транспорта глюкозы через мембрану полиморфно-ядерных лейкоцитов. Согласно существующим предположениям, микросомальный транспорт глюкозо-6-фосфата играет определенную роль в антиоксидантной защите нейтрофилов, а генетическая поломка транслоказы глюкозо-6-фосфатазы ведет к нарушению клеточных функций, в частности, апоптоза, что может служить объяснением нейтрофильной дисфункции у больных с ГБ Ib типа.
Причиной возникновения ГБ III типа являются мутации гена AGL, кодирующего гликоген-деветвящий фермент, который представляет собой большой мономерный белок с молекулярной массой приблизительно 160 кД и имеет 2 каталитических единицы: амило-1, 6-глюкозидазу и , способные функционировать независимо друг от друга, однако для нормального действия деветвящего фермента необходима активность обеих каталитических единиц. Подавляющее большинство больных имеют дефицит этого энзима как в печени, так и в мышцах (подтип IIIa), однако примерно у 15% пациентов он отмечается только в печени (подтип IIIb). Наличие указанных подтипов объясняется различной экспрессией фермента в тканях. В редких случаях селективное снижение активности либо амило-1, 6-глюкозидазы, либо приводит к развитию подтипов IIIc и IIId болезни, соответственно. Генный локус ГБ III типа соответствует 1p21.2. Он содержит 35 экзонов, занимающих 85 kb геномной ДНК. Тип наследования — аутосомно-рецессивный.
Наличие указанных подтипов объясняется различной экспрессией фермента в тканях. В редких случаях селективное снижение активности либо амило-1, 6-глюкозидазы, либо приводит к развитию подтипов IIIc и IIId болезни, соответственно. Генный локус ГБ III типа соответствует 1p21.2. Он содержит 35 экзонов, занимающих 85 kb геномной ДНК. Тип наследования — аутосомно-рецессивный.
Патогенез ГБ III типа. Амило-1, 6-глюкозидаза участвует в метаболизме гликогена в точках ветвления гликогенового «дерева». Фермент является бифункциональным, с одной стороны превращая лимит-декстрин в гликоген с наружными цепями нормальной длины и, с другой, освобождая глюкозу путем гидролиза , 6-глюкозидной связи. Мутация фермента сопровождается нарушением высокоспецифического процесса гликогенолиза, приводя к накоплению в тканях (печень, мышцы) молекул гликогена аномальной формы с укороченными наружными цепями, который оказывает цитотоксическое действие, приводя к дисфункции соответствующих органов. Механизмы формирования гипогликемии и лактатацидоза сходны с таковыми при ГБ I типа.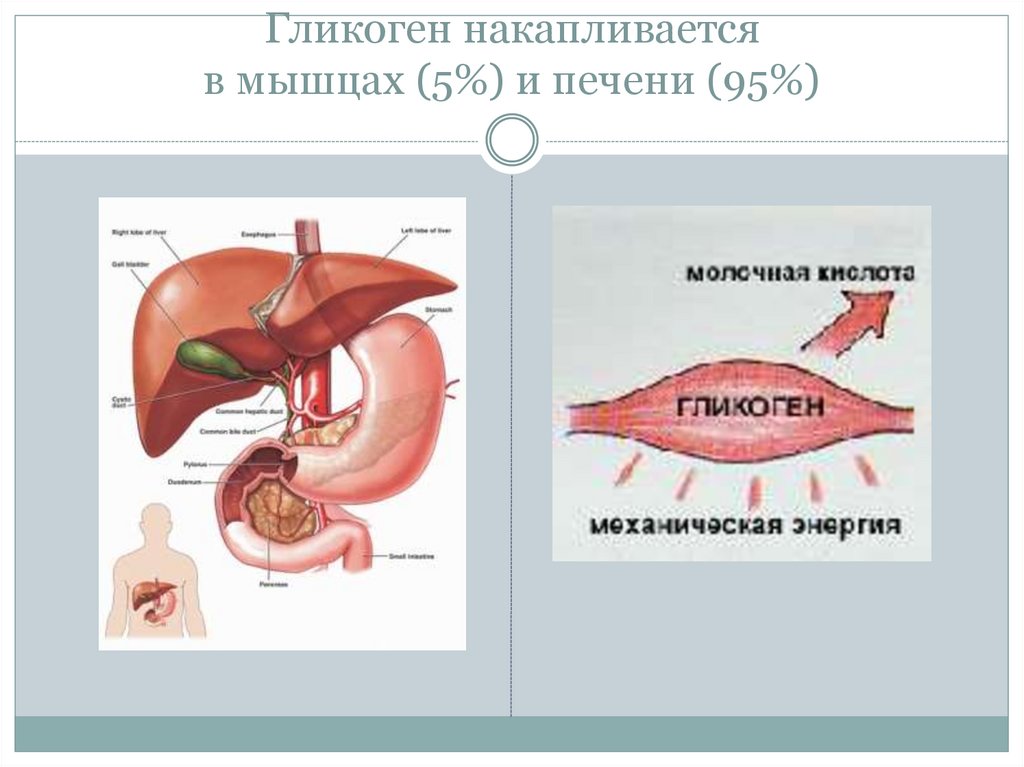
Причиной возникновения ГБ IV типа являются мутации в гене GBE1, кодирующем амило-1, 4:1, 6-глюкантрансферазу, что приводит к ее недостаточности в печени, мышцах, лейкоцитах, эритроцитах и фибробластах. Мутации в этом же гене вызывают полигликозановую болезнь у взрослых. Генный локус ГБ IV типа соответствует 3p12.2. Тип наследования — аутосомно-рецессивный.
Патогенез ГБ IV типа. Амило-1, 4:1, 6-глюкантрансфераза участвует в метаболизме гликогена при точках ветвления гликогенового «дерева». Она соединяет сегмент по крайней мере из шести , 4-сцепленных глюкозидных остатков наружных цепей гликогена с гликогеновым «деревом» связью. Мутация фермента нарушает нормальный синтез гликогена, что приводит к образованию аномального полисахарида с амилопектиноподобной структурой (полигликозгликан). Накапливаясь в различных тканях, включая печень и мышцы, он повреждает их клетки.
Причиной возникновения ГБ VI типа являются мутации в гене PYGL, кодирующего фосфорилазу печени. Генный локус ГБ VI типа соответствует 14q21. 1. Тип наследования — аутосомно-рецессивный.
1. Тип наследования — аутосомно-рецессивный.
Патогенез ГБ VI типа. Дефицит печеночной фосфорилазы, катализирующей первую реакцию распада гликогена, ведет к его избыточному накоплению в гепатоцитах.
Причиной возникновения ГБ IXa типа являются мутации гена PHKB2, кодирующего печеночной киназы фосфорилазы, что приводит к ее недостаточности в печени и эритроцитах (подтип IXa1) или только в печени (подтип IXa2). Генный локус ГБ IXa типа соответствует Xp22.13. Тип наследования — X-сцепленный.
Причиной возникновения ГБ IXb типа являются мутации гена PHKB, кодирующего мышечной/печеночной киназы фосфорилазы. Генный локус ГБ IXb типа соответствует 16q12.1. Тип наследования — аутосомно-рецессивный.
Причиной возникновения ГБ IXc типа являются мутации гена PHKG2, кодирующего тестикулярную/печеночную изоформу киназы фосфорилазы. Генный локус ГБ IXc типа соответствует 16p11.2. Установлено, что ген PHKG2 содержит 10 экзонов и занимает 9,5 kb геномной ДНК. Тип наследования — аутосомно-рецессивный.
Патогенез ГБ IX типа. Дефицит любой из субъединиц киназы фосфорилазы b нарушает процесс фосфоролиза печеночной фосфорилазы b в фосфорилазу a, что ведет к нарушению активации последней и в итоге — к невозможности расщепления гликогена.
Фосфокиназа состоит из 4 разных субъединиц, каждую из которых кодируют различные гены, располагающиеся в различных хромосомах и по-разному экспрессирующиеся в различных тканях: , , , . Субъединицы и выполняют регуляторные функции, функцию, связывания ионов Ca2+. Субъединица имеет две изоформы — мышечную и печеночную, кодируемые двумя разными генами, располагающимися на X-хромосоме. Гены, кодирующие остальные субъединицы, находятся в аутосомных хромосомах. Мутации в генах PHKA2, PHKAB и PHKG2 вызывают ГБ IXa, IXb и IXc типов, соответственно. При развитии одной из четырех основных форм болезни поражаются соответственно, по убыванию частоты встречаемости, печень, печень и мышцы, только мышцы, только сердце. Имеется также подтип IXd (мышечная форма), вызываемый мутациями в гене PHKA1.
Причиной возникновения ГБ 0 типа являются мутации в генах GYS2 и GYS2, кодирующих печеночную и мышечную гликогенсинтазу, соответственно. Генный локус ГБ 0 типа соответствует: 12p12.1 — для печеночной формы, 19q13.33 — для мышечной. Ген GYS2 содержит 16 экзонов и занимает более 30 kb геномной ДНК. Тип наследования — аутосомно-рецессивный.
Патогенез ГБ 0 типа. При отсутствии гликогенситназы невозможен адекватный синтез гликогена, что приводит к выраженному снижению его содержания в печени или мышцах.
Гликогенозы. Что такое Гликогенозы?
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Гликогенозы – наследственные болезни, в основе которых лежит генетический дефект производства ферментов, принимающих участие в метаболизме углеводов. Характерный общий признак – чрезмерное отложение гликогена в миоцитах, гепатоцитах и других клетках организма. Гликогенозы проявляются симптомами гипогликемии, гепатомегалии, мышечной слабости, печеночной, сердечной, дыхательной и почечной недостаточности. Диагностика включает биохимический анализ крови, морфологическое исследование биопсийного материала мышц и печени, определение активности ферментов, молекулярно-генетические тесты. Лечение основано на лечебном питании, медикаментозной коррекции метаболических расстройств, в ряде случаев требуются операции.
Характерный общий признак – чрезмерное отложение гликогена в миоцитах, гепатоцитах и других клетках организма. Гликогенозы проявляются симптомами гипогликемии, гепатомегалии, мышечной слабости, печеночной, сердечной, дыхательной и почечной недостаточности. Диагностика включает биохимический анализ крови, морфологическое исследование биопсийного материала мышц и печени, определение активности ферментов, молекулярно-генетические тесты. Лечение основано на лечебном питании, медикаментозной коррекции метаболических расстройств, в ряде случаев требуются операции.
МКБ-10
E74.0 Болезни накопления гликогена
- Причины гликогенозов
- Патогенез
- Классификация
- Симптомы гликогенозов
- Осложнения
- Диагностика
- Лечение гликогенозов
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Исследование гликогенозов ведется с 1910 года. В 1928-29 годах была описана симптоматика гликогеноза I типа – «болезни накопления гликогена». Лишь в 1952 году удалось выявить ферментный дефект и установить его связь с развитием симптомов. Патогенетические механизмы и способы лечения до сих пор остаются не до конца изученными. К настоящему времени выделено 12 типов гликогенозов, наиболее полно исследовано 9. Распространенность низкая, в среднем составляет 1 случай на 40-68 тысяч населения. Эпидемиологические показатели одинаковы среди представителей обоих полов, но при X-рецессивном наследовании мужчины болеют чаще. Симптомы проявляются в период новорожденности или в раннем детстве, течение чаще непрерывно прогрессирующее.
В 1928-29 годах была описана симптоматика гликогеноза I типа – «болезни накопления гликогена». Лишь в 1952 году удалось выявить ферментный дефект и установить его связь с развитием симптомов. Патогенетические механизмы и способы лечения до сих пор остаются не до конца изученными. К настоящему времени выделено 12 типов гликогенозов, наиболее полно исследовано 9. Распространенность низкая, в среднем составляет 1 случай на 40-68 тысяч населения. Эпидемиологические показатели одинаковы среди представителей обоих полов, но при X-рецессивном наследовании мужчины болеют чаще. Симптомы проявляются в период новорожденности или в раннем детстве, течение чаще непрерывно прогрессирующее.
Гликогенозы
Причины гликогенозов
Единственным фактором, провоцирующим развитие гликогеновых болезней, является генетический дефект, в результате которого возникает недостаточность определенного фермента, участвующего в обмене глюкозы. Все гликогенозы за исключением IX типа наследуются по аутосомно-рецессивному принципу.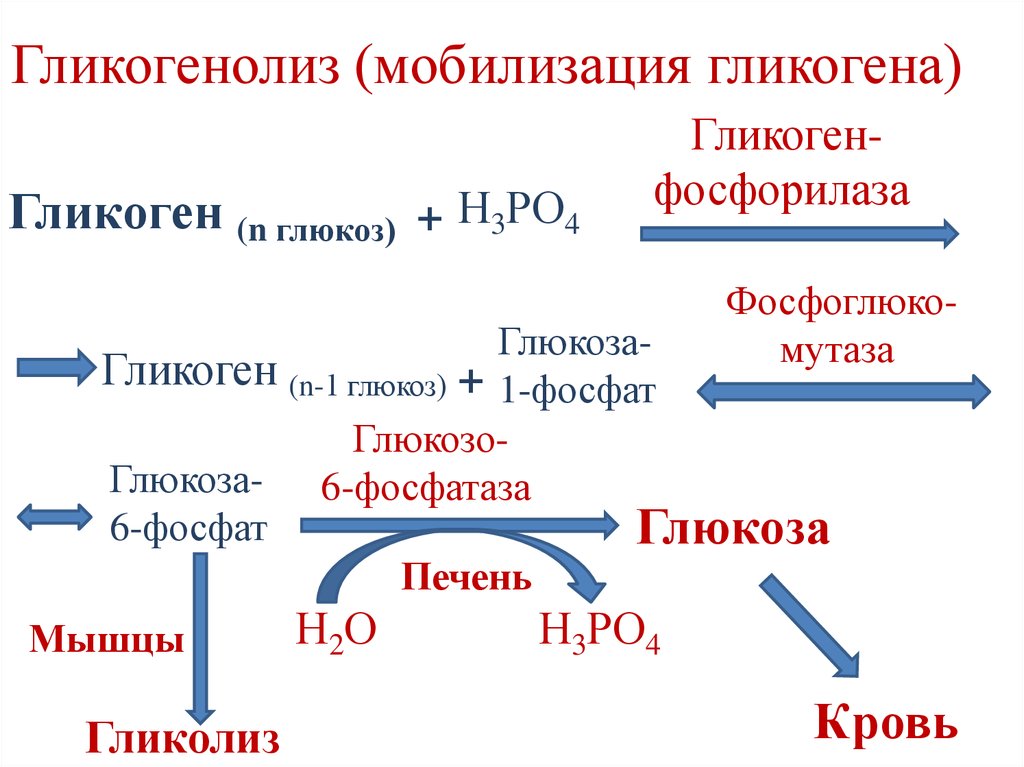 Это означает, что мутационный ген расположен на хромосоме, не сцепленной с полом, проявление заболевания возможно только при наследовании мутаций от каждого из родителей – при наличии двух рецессивных измененных генов в аллели. Если дефектным является один ген из пары, то другой – доминантный, нормальный – обеспечивает организм достаточным количеством фермента. Человек при этом становится носителем гликогеноза, но не болеет. В парах, где оба партнера – носители, вероятность рождения больного ребенка составляет 25%. При гликогенозе типа IX патологический ген локализован в половой X-хромосоме. Гемизиготные мужчины имеют пару XY, всегда больны гликогенозом, передают дефект всем своим дочерям. Вероятность передачи мутации от женщины-носительницы детям обоих полов составляет 50%.
Это означает, что мутационный ген расположен на хромосоме, не сцепленной с полом, проявление заболевания возможно только при наследовании мутаций от каждого из родителей – при наличии двух рецессивных измененных генов в аллели. Если дефектным является один ген из пары, то другой – доминантный, нормальный – обеспечивает организм достаточным количеством фермента. Человек при этом становится носителем гликогеноза, но не болеет. В парах, где оба партнера – носители, вероятность рождения больного ребенка составляет 25%. При гликогенозе типа IX патологический ген локализован в половой X-хромосоме. Гемизиготные мужчины имеют пару XY, всегда больны гликогенозом, передают дефект всем своим дочерям. Вероятность передачи мутации от женщины-носительницы детям обоих полов составляет 50%.
Патогенез
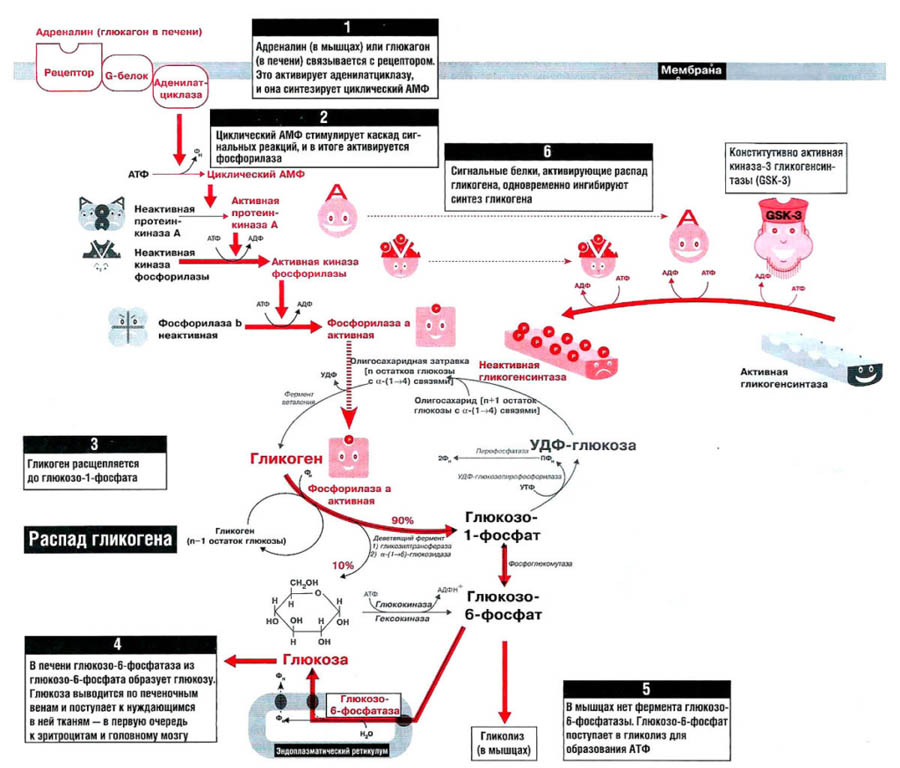
Патогенетическая основа всех гликогенозов – невозможность процесса синтеза и распада гликогена, его накопление в тканях. Гликоген является единственным резервным полисахаридом организма, своеобразным энергетическим «депо» – после приема пищи излишек глюкозы превращается в гликоген печени и мышц, затем постепенно расщепляется обратно до глюкозы.
При гликогеновых болезнях типов 1-11 возникает генетически обусловленная недостаточность какого-либо фермента, катализирующего цепочку глюкоза-гликоген-глюкоза. 1 тип характеризуется дефектом глюкозо-6-фосфатазы и глюкозо-6-фосфаттранслоказы, 2 тип – альфа-1,4-глюкозидазы, 3 тип – амило-1,6-глюкозидазы, 4 тип – D-1,4-глюкано-α-глюкозилтрансферазы, 5 тип – гликогенфосфорилазы миоцитов, 6 тип – крахмалфосфорилазы гепатоцитов, 7 тип – фосфоглюкомутазы, 8 тип – фосфофруктомутазы, 9 тип – киназы фосфорилазы гепатоцитов. Из-за сниженной активности или полного отсутствия фермента гликоген накапливается в мышцах, печени, редко – в других тканях. Изменяется структура и функциональность органов, развиваются различные формы органной недостаточности.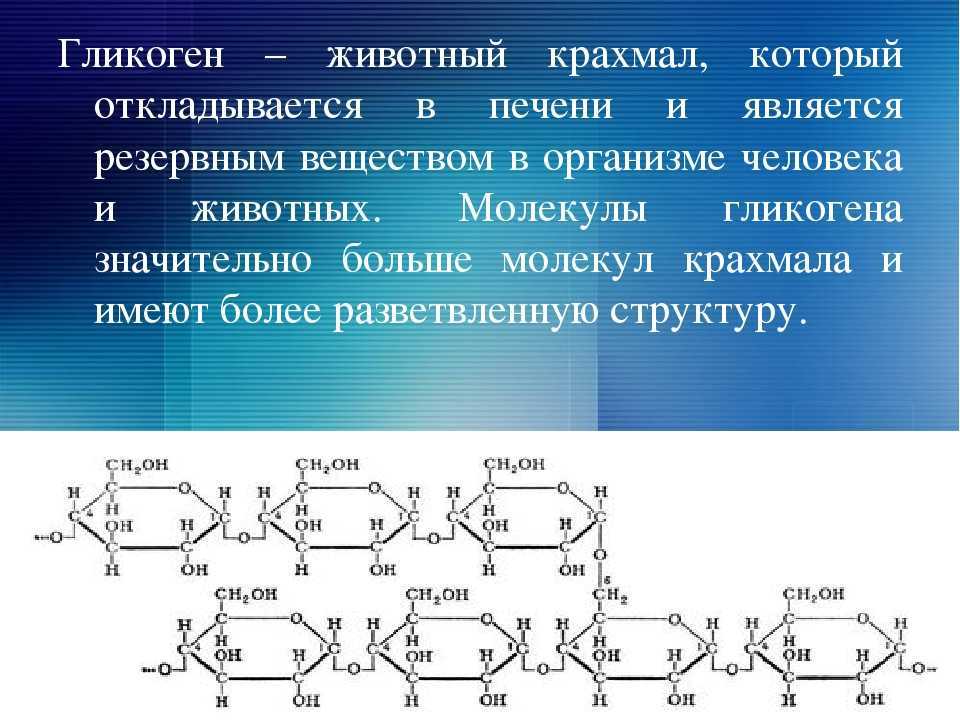
Классификация
С учетом ферментативного дефекта и особенностей клинических проявлений выделяют 12 вариантов гликогенозов, от 0 до XI. Кроме того, описаны случаи комбинированных типов, когда определяется дефицит двух ферментов, а также случаи неидентифицируемых типов, при которых выделить ферментный дефект не удается. Согласно ведущему патогенетическому механизму гликогеновые болезни подразделяются на три больших группы:
- Печеночные. Включают гликогенозы всех типов, кроме II, V и VII. Гликоген откладывается преимущественно в гепатоцитах. Характерна гепатомегалия, гипогликемия через 2 часа после поступления углеводов. При I типе заболевания также поражаются почки, при III и IV типах развиваются миопатии.
- Мышечные. В данную группу входят болезни типов VII и V. Изменена ферментативная активность в мышечной ткани, нарушено энергообеспечение мышц. Типичные симптомы – миалгии, судороги.
- Смешанные. Гликогеноз II типа отличается тем, что в патологический процесс вовлекаются все гликогенсодержащие ткани.
 Гликоген скапливается в лизосомах и цитоплазме клеток. Страдают многие органы, возрастает риск смерти по причине сердечной или дыхательной недостаточности.
Гликоген скапливается в лизосомах и цитоплазме клеток. Страдают многие органы, возрастает риск смерти по причине сердечной или дыхательной недостаточности.
Симптомы гликогенозов
Агликогеноз развивается в периоде новорожденности либо раннего детства. Низкое содержание гликогена в печени проявляется резко выраженной гипогликемией натощак. Наблюдается заторможенность, глубокий сон, потеря сознания, бледность кожи, тошнота, рвота, судороги ночью и в утренние часы. Внешне пациенты низкорослые, имеют пониженную плотность костной ткани, повышенный риск переломов. При болезни Гирке (I тип) симптомы дебютируют в первые 4 месяца жизни. Характерен плохой аппетит, приступы рвоты, недостаток веса, увеличение печени, диспропорциональность строения тела – круглое лицо, большой живот, тонкие конечности.
Клинические признаки болезни Помпе (II тип) определяются в течение нескольких недель после рождения. Дети вялые, малоподвижные, с ослабленным сосательным рефлексом, сниженным аппетитом.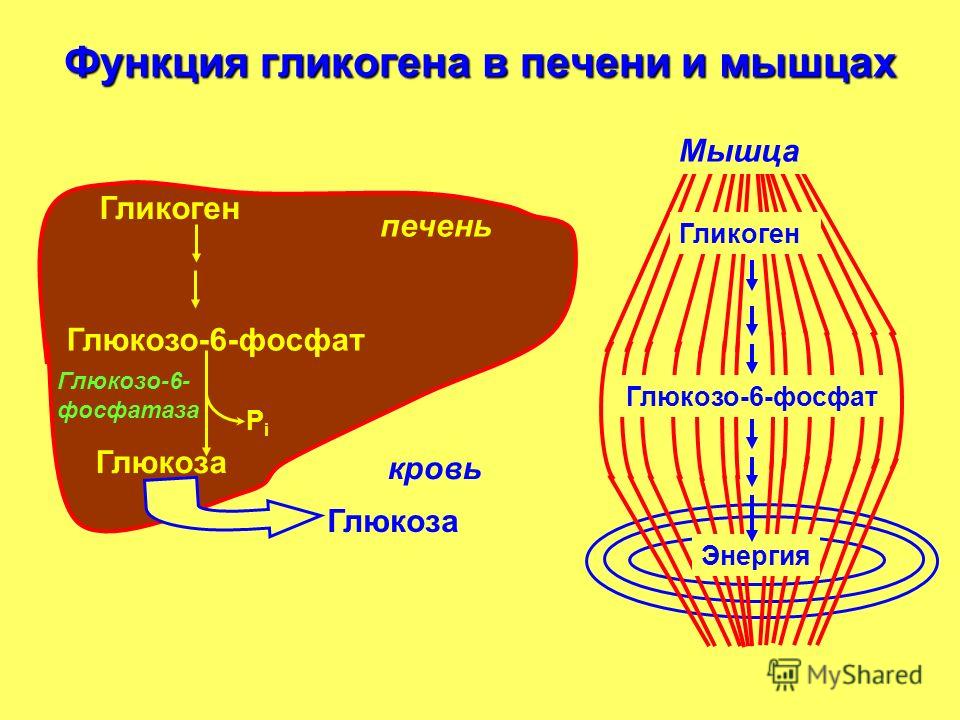 Гепатомегалия изменяет пропорции тела – живот увеличивается, руки и ноги остаются тонкими. Поражается сердце, легкие, нервная система. Высок риск сердечной и легочной недостаточности. У пациентов с болезнью Форбса (III тип) симптомы слабой и умеренной выраженности. На первый план выходит гипогликемия постабсорбционного периода, гепатомегалия, накопление подкожного жира в области туловища. Ведущие симптомы болезни Андерсена (IV тип)– мышечная слабость, плохая переносимость физической нагрузки, судороги.
Гепатомегалия изменяет пропорции тела – живот увеличивается, руки и ноги остаются тонкими. Поражается сердце, легкие, нервная система. Высок риск сердечной и легочной недостаточности. У пациентов с болезнью Форбса (III тип) симптомы слабой и умеренной выраженности. На первый план выходит гипогликемия постабсорбционного периода, гепатомегалия, накопление подкожного жира в области туловища. Ведущие симптомы болезни Андерсена (IV тип)– мышечная слабость, плохая переносимость физической нагрузки, судороги.
Болезнь Томсона представлена гепатомегалией, нистагмом, атаксией, прогрессирующими неврологическими нарушениями с мышечной гипертонией, децеребрацией. Типичные проявления болезни Мак-Ардля (V тип) – боли, спазматические сокращения, чрезмерная утомляемость и слабость мышц даже после незначительной нагрузки. Иногда тонические судороги переходят в генерализованные, что сопровождается общей скованностью. Проявления болезни Герса (VI тип) менее выраженные, пациенты способны переносить легкие и умеренные физические нагрузки, не испытывая дискомфорта.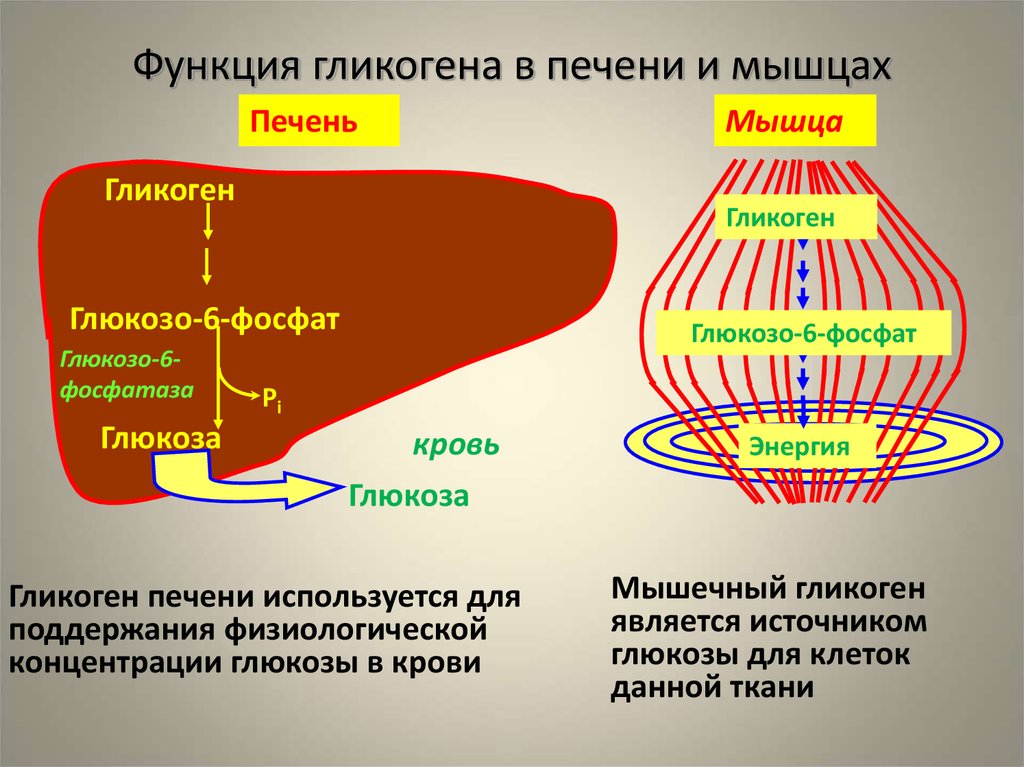 Дополнительно обнаруживаются признаки поражения печени – угнетение аппетита, рвота, тошнота, боли в правом боку.
Дополнительно обнаруживаются признаки поражения печени – угнетение аппетита, рвота, тошнота, боли в правом боку.
Течение болезни Таруи (VII тип) включает непереносимость физической нагрузки, сопровождающуюся тошнотой и рвотой, болезненными спазмами мышц. Поступление глюкозы не повышает способность совершать физические действия. После употребления пищи симптомы обостряются. Наиболее мягкое течение свойственно болезни Хага (IX тип). У больных детей увеличивается печень, задерживается моторное развитие и рост, формируется мышечная гипотония. С возрастом симптомы самостоятельно редуцируются. Гликогеноз X типа крайне редок, характеризуется гепатомегалией, при длительном течении снижается переносимость физических нагрузок. Гликогеноз XI типа сопровождается значительным увеличением печени, задержкой роста и физического развития, рахитом. У подростков нередко наблюдается сокращение объема печени, ускорение роста.
Осложнения
При разновидностях гликогенозов, сопровождающихся гипогликемией, существует риск развития гипогликемической комы.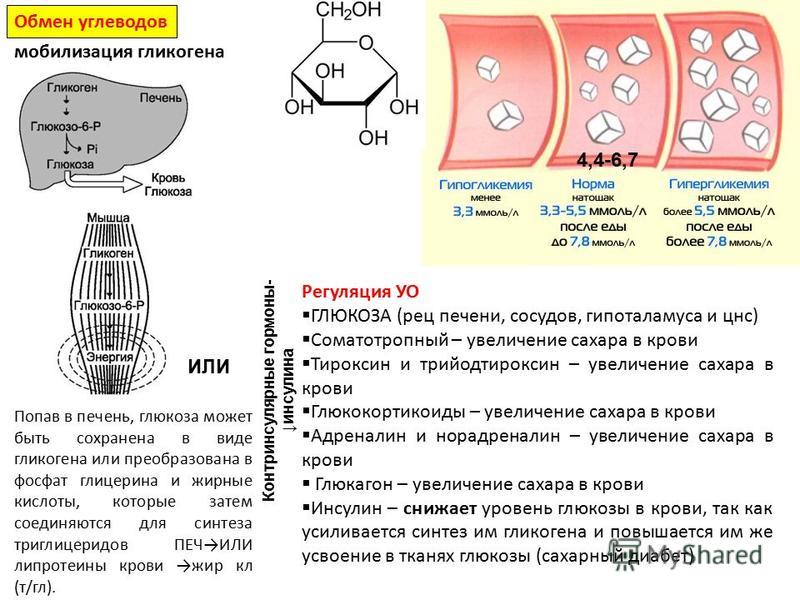
Диагностика
При подозрении на гликогеноз ребенку рекомендуется консультация врача-генетика, педиатра, гастроэнтеролога, гепатолога. В первую очередь специалист собирает анамнез, проводит клинический опрос и осмотр. Поскольку заболевание передается аутосомно-рецессивным способом, семейные случаи выявляют редко. Распространены жалобы на слабость, апатичность ребенка, бледность и желтушность кожи, отказ от еды или повышенный аппетит, трудности пробуждения утром, тремор, судороги. При осмотре врач отмечает увеличение размера печени, выпирание живота, задержку роста, мышечную гипотрофию, специфическое отложение подкожной жировой клетчатки, ксантомы.
- Биохимическое исследование крови. По результатам анализа обнаруживается гипогликемия с уровнем глюкозы натощак 0,6-3 ммоль/л, лактатацидоз с концентрацией молочной кислоты 3-10 ммоль/л (кроме гликогеноза 4 типа). Дополнительно выявляется увеличение показателей триглицеридов, общего холестерина, ЛПНП, ЛПОНП, мочевой кислоты, печеночных ферментов.
- Исследование биоптата печени, мышц. При изучении ткани печени наиболее распространенными характеристиками являются повышенное количество гликогена и его глыбчатое распределение в цитоплазме гепатоцитов, иногда – в вакуолизированных ядрах. Определяется выраженная белковая и/или крупно- и мелкокапельная жировая дистрофия гепатоцитов, их некроз, ограниченные очаги фиброза в местах гибели клеток.
 Возможны признаки цирроза. При мышечных типах болезней исследуется мышечный биоптат, в котором просматриваются субсарколеммальные накопления структурно нормального гликогена.
Возможны признаки цирроза. При мышечных типах болезней исследуется мышечный биоптат, в котором просматриваются субсарколеммальные накопления структурно нормального гликогена. - Исследование ферментов. Активность ферментов изучается в культуре кожных фибробластов, биоптате мышечной и печеночной ткани, лейкоцитах. При гликогенозах с хроническим медленно прогрессирующим течением снижение функциональности фермента легкое или умеренное. При тяжелом течении фермент отсутствует либо его активность минимальна.
- УЗИ брюшной полости. Отмечается выраженное увеличение печени, особенно левой ее доли. Характерна гиперэхогенность и структурная диффузная неоднородность паренхимы (множественные мелкие гиперэхогенные эхосигналы, распределенные равномерно). В дистальных отделах паренхимы прохождение ультразвука ослаблено. Возможно обнаружение структурно разнообразных печеночных аденом, увеличение размеров почек, селезенки и поджелудочной железы.
Комплекс диагностических исследований подбирается индивидуально в зависимости от возраста пациента и предполагаемого типа гликогеновой болезни. Может потребоваться молекулярно-генетическая диагностика (секвенирование генов с целью выявления мутации), электромиография, ЭХО-КГ, ОАК, коагулограмма.
Может потребоваться молекулярно-генетическая диагностика (секвенирование генов с целью выявления мутации), электромиография, ЭХО-КГ, ОАК, коагулограмма.
Лечение гликогенозов
Специфические методы терапии не разработаны. Патогенетическое лечение проводится консервативно, направлено на устранение гипогликемии, метаболического ацидоза, кетоза, гиперлипидемии, коррекцию дисфункции гепатобилиарного комплекса и желудочно-кишечного тракта. При развитии осложнений (серьезном поражении внутренних органов) выполняются хирургические операции. Медицинская помощь пациентам включает следующие направления:
- Диетотерапию. Для минимизации метаболических нарушений составляется индивидуальный план питания. Больным рекомендуется снизить количество жиров, сахарозы, фруктозы и галактозы для уменьшения гиперлипидемии и ацидоза. При первом типе гликогеноза назначается диета с увеличенным потреблением углеводов. В частности, показано употребление сырого кукурузного крахмала с медленной усвояемостью, позволяющей предупредить гипогликемию.
 При типах 3, 4 и 9 вводится рацион с преобладанием животного белка и дробным питанием.
При типах 3, 4 и 9 вводится рацион с преобладанием животного белка и дробным питанием. - Лекарственную коррекцию симптомов. В рамках комплексного лечения применяется кокарбоксилаза для увеличения производства ацетилкофермента А, кортикостероидов и глюкагона для стимуляции глюконеогенеза. Дефицит карнитина компенсируется левокарнитином. При вторичных тубулопатиях, печеночных и билиарных дисфункциях используются желчегонные препараты, гепатопротекторы, липотропные вещества. При признаках ацидоза показаны щелочные растворы внутривенно. При почечной дисфункции, протеинурии – ингибиторы АПФ. При гиперурикемии – урикодепрессоры. При нейтропении – гранулоцитарный колониестимулирующий фактор.
- Хирургическое лечение. Пациентам с тяжелыми фатальными поражениями печени может потребоваться ортотопическая трансплантация органа. Показанием к операции является цирроз с осложнениями, часто развивающийся при третьем и четвертом типе патологии. В отдельных случаях хирургическое вмешательство целесообразно при аденомах печени с высоким риском трансформации в злокачественную опухоль.
 Трансплантация почек иногда выполняется больным с хронической почечной недостаточностью.
Трансплантация почек иногда выполняется больным с хронической почечной недостаточностью.
Прогноз и профилактика
Эффективность терапии, вероятность осложнений и летального исхода зависят от типа патологии. Одни гликогенозы незначительно ухудшают качество жизни больных, компенсируются по мере взросления, другие – не поддаются лечению и неизбежно завершаются смертью. Для снижения риска рождения ребенка с гликогенозом супружеским парам из группы риска – имеющим семейный отягощенный анамнез, детей с подтвержденным диагнозом – требуется медико-генетическое консультирование, пренатальная диагностика.
Вы можете поделиться своей историей болезни, что Вам помогло при лечении гликогенозов.
Источники
- Педиатрия. Национальное руководство в 2 томах. Том 2./ под ред. Баранова А.А. — 2009.
- Гликогеновая болезнь у детей. Клинические рекомендации/ Союз педиатров России – 2016.
- Заболевания печени и желчных путей/ Шерлок Ш.
 Дули Дж. — 1999.
Дули Дж. — 1999. - Настоящая статья подготовлена по материалам сайта: https://www.krasotaimedicina.ru/
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Метаболизм гликогена в печени во время и после длительных упражнений на выносливость
- Home
- Метаболизм гликогена в печени во время и после длительных упражнений на выносливость
 Однако, когда нетренированные люди участвуют в физических упражнениях, они метаболизируют гликоген в печени намного быстрее, чем их тренированные коллеги. Они ссылаются на одно исследование, которое показало, что у нетренированных людей скорость гликогенолиза печени (распада гликогена) составляет 6,9.мг/кг/мин, в то время как у тренированных людей скорость составляла 5,3 мг/кг/мин. Это означает, что во время упражнений средней и высокой интенсивности нетренированные люди истощали запасы гликогена в печени и испытывали усталость через 118 минут, в то время как запасы тренированных людей не истощались до 153 минут. Авторы приходят к выводу, что «более низкая скорость гликогенолиза в печени при тренировке на выносливость, вероятно, способствует большей выносливости/возможностям, способствуя поддержанию (высокой) скорости окисления углеводов и гомеостаза глюкозы в крови на последних этапах упражнений».
Однако, когда нетренированные люди участвуют в физических упражнениях, они метаболизируют гликоген в печени намного быстрее, чем их тренированные коллеги. Они ссылаются на одно исследование, которое показало, что у нетренированных людей скорость гликогенолиза печени (распада гликогена) составляет 6,9.мг/кг/мин, в то время как у тренированных людей скорость составляла 5,3 мг/кг/мин. Это означает, что во время упражнений средней и высокой интенсивности нетренированные люди истощали запасы гликогена в печени и испытывали усталость через 118 минут, в то время как запасы тренированных людей не истощались до 153 минут. Авторы приходят к выводу, что «более низкая скорость гликогенолиза в печени при тренировке на выносливость, вероятно, способствует большей выносливости/возможностям, способствуя поддержанию (высокой) скорости окисления углеводов и гомеостаза глюкозы в крови на последних этапах упражнений». К счастью, гликоген легко пополняется и поддерживается во время и после тренировки.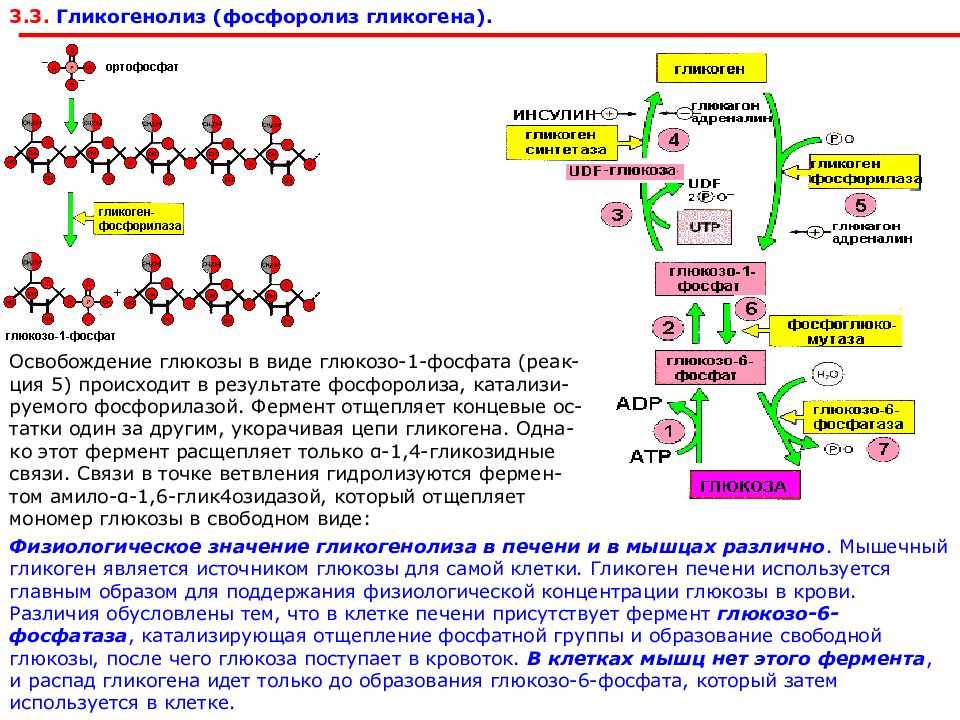 Гонсалес и др. ссылаются на ряд исследований, которые предполагают, что прием углеводов, особенно глюкозы или сахарозы (глюкоза-фруктоза) во время физических упражнений, может уменьшить истощение гликогена в печени. Считается, что потребление 1,2 г/кг углеводов во время восстановления идеально подходит для быстрого насыщения.
Гонсалес и др. ссылаются на ряд исследований, которые предполагают, что прием углеводов, особенно глюкозы или сахарозы (глюкоза-фруктоза) во время физических упражнений, может уменьшить истощение гликогена в печени. Считается, что потребление 1,2 г/кг углеводов во время восстановления идеально подходит для быстрого насыщения.
Хотя не было доказано, что фруктоза ускоряет восполнение запасов гликогена в мышцах после тренировки, она увеличивает восполнение запасов гликогена в печени. Гонсалес и др. обратите внимание на многочисленные исследования, показывающие, что совместное употребление фруктозы с глюкозой после тренировки может почти удвоить скорость восполнения запасов гликогена в печени независимо от общего объема потребляемых углеводов. Кроме того, комбинации фруктозы и глюкозы лучше переносятся и вызывают меньший желудочно-кишечный (ЖКТ) стресс, чем глюкоза в отдельности.
В заключение, примерно через 90 минут умеренно-высокой интенсивности запасы гликогена в печени истощаются. Употребление углеводов, глюкозы или сахарозы во время тренировки может ослабить истощение. Для восстановления после тренировки рекомендуются комбинации глюкоза-фруктоза или глюкоза-галактоза для быстрого восполнения и толерантности к желудочно-кишечному тракту.
Употребление углеводов, глюкозы или сахарозы во время тренировки может ослабить истощение. Для восстановления после тренировки рекомендуются комбинации глюкоза-фруктоза или глюкоза-галактоза для быстрого восполнения и толерантности к желудочно-кишечному тракту.
Метаболизм гликогена в печени во время и после длительных упражнений на выносливость впервые появился на FructoseFacts.
У вас есть вопросы о низкокалорийных подсластителях? Хотите узнать больше о ведении здорового образа жизни? Вы просили, и мы слушали. Наши постоянные зарегистрированные диетологи ответили на самые популярные вопросы о низкокалорийных подсластителях.
Контакты для СМИ
CCC Staff
[email protected]
Болезнь накопления гликогена | Boston Children’s Hospital
Болезнь накопления гликогена (GSD) — это название группы заболеваний, которые нарушают способность организма вырабатывать гликоген или преобразовывать гликоген в глюкозу. В зависимости от типа GSD у ребенка гликоген может накапливаться в печени, мышцах или и там, и там. GSD также может поражать клетки крови, сердце, почки и другие органы.
В зависимости от типа GSD у ребенка гликоген может накапливаться в печени, мышцах или и там, и там. GSD также может поражать клетки крови, сердце, почки и другие органы.
Гликоген является основным источником энергии в организме. В норме гликоген хранится в печени до тех пор, пока организму не потребуется энергия. Затем ферменты превращают гликоген в глюкозу, чтобы она могла перемещаться по кровотоку к клеткам, которым требуется топливо. Каждая клетка тела содержит ферменты, но у детей с GSD отсутствует один из ферментов, отвечающих за выработку гликогена или превращение гликогена в глюкозу.
GSD — редкое заболевание. По данным Национальной организации редких заболеваний, GSD поражает менее 1 из 40 000 человек в Соединенных Штатах.
Какие существуют типы GSD?
Существует множество различных типов GSD, в зависимости от того, какой фермент отсутствует. Некоторые типы поражают только печень, другие только мышцы, а некоторые поражают и печень, и мышцы. Каждый тип имеет немного разные симптомы.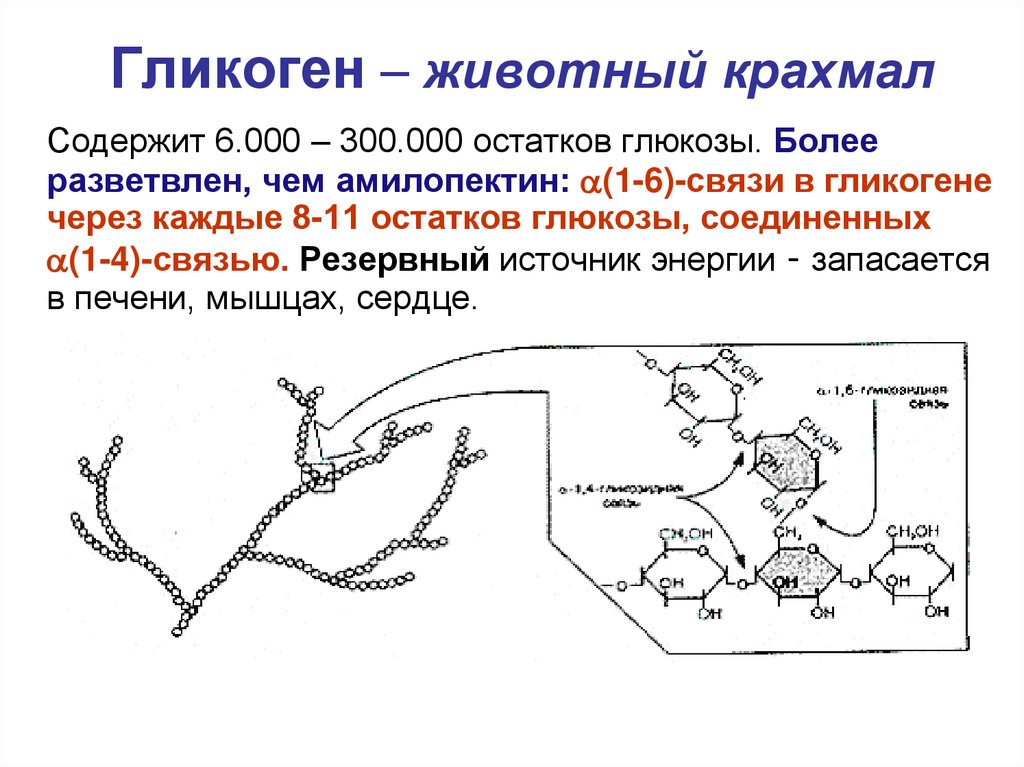 Лечение различается для различных типов GSD.
Лечение различается для различных типов GSD.
Наиболее распространенные типы GSD включают:
Болезнь накопления гликогена типа I (GSD I) , также известную как болезнь фон Гирке, составляет около 25 процентов всех детей с GSD. Симптомы обычно появляются, когда ребенку от 3 до 4 месяцев, и могут включать гипогликемию (низкий уровень сахара в крови), которая может вызывать усталость, постоянный голод и раздражительность. Печень и иногда почки отекают из-за накопления гликогена.
Болезнь накопления гликогена III типа (GSD III) , также известная как болезнь Кори или болезнь Форбса, вызывает накопление гликогена в печени и мышцах. Симптомы обычно появляются в течение первого года жизни. У детей с этим типом GSD может быть вздутие живота, задержка роста и слабость мышц.
Болезнь накопления гликогена IV типа (GSD IV) , также известная как болезнь Андерсена, является одним из наиболее серьезных типов GSD. Симптомы обычно появляются в первый месяц жизни ребенка и включают неспособность набирать вес или расти с ожидаемой скоростью.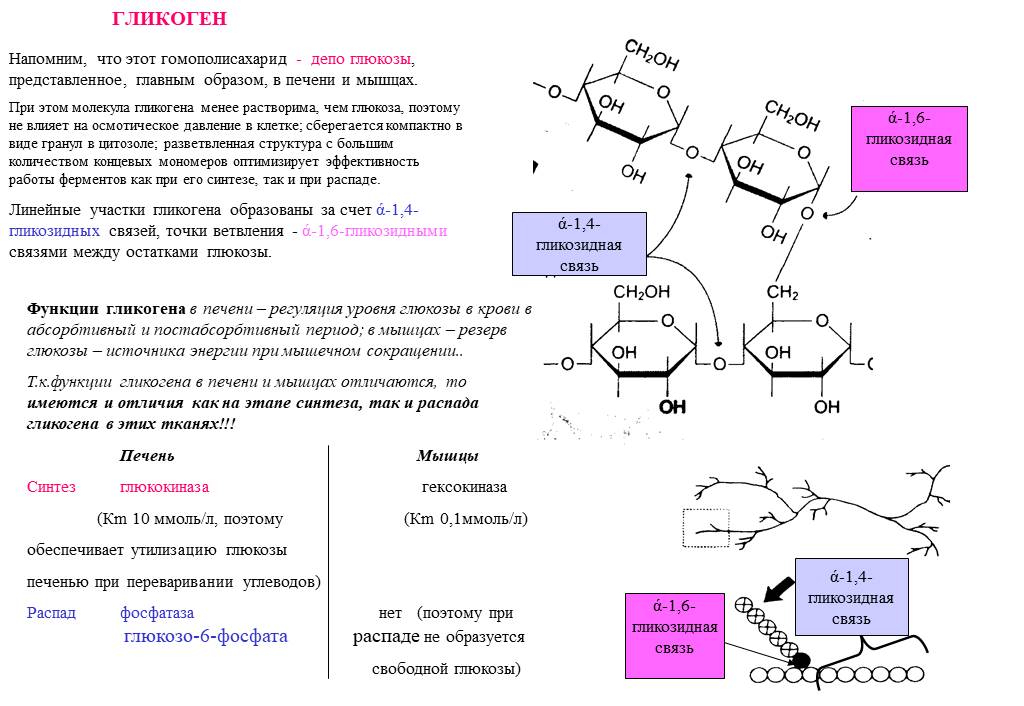 Этот тип GSD часто приводит к циррозу печени, а также может поражать сердце и другие органы. Исходы ребенка зависят от формы GSD IV, которую они унаследовали.
Этот тип GSD часто приводит к циррозу печени, а также может поражать сердце и другие органы. Исходы ребенка зависят от формы GSD IV, которую они унаследовали.
Каковы риски GSD?
Каждый тип GSD несет определенные риски.
У младенцев с типом I (GSD I) может быть низкий уровень сахара в крови. Этот тип GSD также может привести к лактоацидозу, накоплению молочной кислоты, что может вызвать болезненные мышечные спазмы. По мере взросления у детей с GSD I может наблюдаться задержка полового созревания и слабость костей (остеопороз). Другие риски включают:
- подагру, тип артрита
- аденомы, опухоли печени, обычно доброкачественные (нераковые)
- воспалительное заболевание кишечника (тип 1b)
- проблемы с зубами
- рецидивирующие инфекции (тип 1b)
- легочная гипертензия
Младенцы с типом III (GSD III) могут иметь низкий уровень сахара в крови и избыток жира в крови. По мере взросления их печень может увеличиваться.